










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.30 no.3 Bogotá July/Sept. 2005
Inflamación, tejido adiposo, resistencia a la insulina y aterogénesis se expande el rompecabezas
Inflammation, adipose tissue, insulin resistance and atherogenesis the puzzle grows
Dr. Guido Lastra G.: Internista, Endocrinólogo;
Dra. Camila M. Manrique A.: Internista Endocrinóloga;
Dr. Guido Lastra L.: Profesor Titular, Unidad de Endocrinnnología, Facultad de Medicina Universidad Nacional de Colombia. Bogotá, D.C.
Correspondencia Dr. Guido Lastra L, MD. D.Sc, Unidad de Endocrinología, Facultad de Medicina, Universidad Nacional de Colombia. Bogotá D.C.
Recibido: 13/04/05 Aprobado: 03/08/05
Resumen
Durante las últimas décadas la obesidad ha pasado de ser un problema puramente estético, con poca repercusión fisiopatológica, a ser una verdadera epidemia que afecta más de un tercio de las población occidental, y que comienza a afectar cada día más a las generaciones jóvenes. La obesidad se caracteriza por una miríada de alteraciones metabólicas, en las cuales hay una participación clave del tejido adiposo, que se constituye en uno de los órganos endocrinos más complejos y fascinantes descubiertos en los últimos tiempos. El tejido adiposo, vital para la conservación de la energía necesaria para soportar el metabolismo de la economía, coadyuvar en funciones inmunológicas y reproductivas, es afectado por influencias genéticas y ambientales que lo tornan disfuncional. Predomina en la obesidad el tejido adiposo visceral, caracterizado por cambios morfológicos y funcionales que los convierten en fuente de citoquinas, conocidas en la actualidad como adipoquinas, que conducen a un estado inflamatorio crónico de bajo grado. La repercusión de estas alteraciones se refleja en resistencia a la insulina, lesión endotelial y finalmente aterogénesis, que conducen a complicaciones metabólicas y cardiovasculares crónicas, tales como diabetes mellitus 2, enfermedad hipertensiva, enfermedad vascular periférica y enfermedad coronaria, condiciones que constituyen las principales causas de morbimortalidad en el mundo moderno. Paradójicamente, el mundo industrializado parece haber transformado uno de los sistemas orgánicos más antiguos, que permitió la supervivencia del ser humano en difíciles circunstancias de conservación de energía y de lucha contra infecciones generalmente letales, en el origen de las enfermedades que con mayor frecuencia nos afectan en la actualidad. El descubrimiento de los mecanismos íntimos de regulación y disfunción del tejido adiposo, de su relación con la aterogénesis y la enfermedad cardiovascular ayudarán en un futuro a desarrollar estrategias de manejo de la obesidad y del cortejo funerario que la acompaña.
Palabras clave: inflamación, adipocito, resistencia a la insulina, aterogenesis.
Abstract
Over the last few decades, from being a purely esthetic problem without any pathological implications, obesity has become a true epidemic affecting more than a third of the Western population, involving an increasing number of people among the younger generations. Obesity is characterized by a myriad of metabolic derangements, in which adipose tissue plays a key role and has shown to be one of the most complex and fascinating endocrine organs discovered recently. Adipose tissue, whose essential role is to store the energy required to support metabolism and contribute to immunologic and reproductive functions, is compromised by genetic and environmental influences, which turn it into a dysfunctional tissue.
Visceral adipose tissue in obesity is characterized by morphologic and functional changes that turn it into a source of cytokines directly produced by adipocytes. These are currently known as adipokynes, and lead to a chronic, low-grade inflammatory state. These changes induce insulin resistance, endothelial dysfunction and, ultimately, atherosclerosis, which leads to metabolic and cardiovascular complications including type-2 diabetes mellitus, hypertensive disease, peripheral vascular disease and coronary heart disease. These conditions constitute the main causes of morbidity and mortality in the modern Western world. Paradoxically, the industrialized world has transformed one of the oldest organic systems that allowed human beings to survive in difficult circumstances when there was a need to preserve energy and fight against lethal infections, into the very source of the diseases that most commonly affect us currently. The discovery of the intimate mechanisms of regulation and dysfunction of adipose tissue, and of its relationship with atherosclerosis and cardiovascular disease, will contribute to the future development of management strategies for obesity and its accompanying lethal consequences.
Key words: inflammation, adipocyte, insulin resistance, atherogenesis.
Introducción
Las estadísticas son irrefutables: más de 30% de adultos norteamericanos padecen de obesidad, y se calcula que el exceso de peso y las complicaciones metabólicas que la acompañan afectan más de la mitad de los adultos en Estados Unidos (1). La prevalencia de la obesidad ha aumentado en 110% al compararla con la década de los años setenta (2). Una de ellas es la diabetes mellitus tipo 2 (DM2), cuya incidencia aumenta en forma sostenida, a pesar de múltiples esfuerzos de salud pública en todo el mundo industrializado (1). Las complicaciones crónicas diabéticas macrovasculares, en particular la enfermedad coronaria, se caracterizan por aterosclerosis progresiva y, a pesar de ser reducidas, no son del todo abolidas por el control aislado de las cifras de glicemia (3). Si bien ha habido notables avances en el conocimiento de la fisiopatología de la DM2, en particular acerca de la resistencia a la insulina y la falla de las células b del páncreas, aún no se dispone de una teoría que unifique el desarrollo de la enfermedad o de sus complicaciones, con los mecanismos de la aterogénesis.
La formación de placas ateromatosas es la vía final común de un grupo de condiciones aparentemente diferentes en principio, tales como la obesidad, DM2 y la hipertensión arterial (4). Numerosas investigaciones demuestran la participación de mecanismos inflamatorios en el desarrollo de las placas ateroscleróticas (5), desde hace aproximadamente una década han aparecido numerosas publicaciones relacionando inflamación con resistencia a la insulina. Datos recientes muestran cómo obesidad, DM2, hipertensión arterial y enfermedad vascular periférica de origen aterosclerótico están íntimamente asociadas con un estado inflamatorio crónico de bajo grado, y con disfunción endotelial (6).
Evidencia epidemiológica
Desde 1993 existen datos disponibles según los cuales, en pacientes con enfermedad coronaria, los niveles de insulina se correlacionan estadísticamente con aquellos de proteína C reactiva (PCR), un conocido marcador de inflamación. Más recientemente algunas investigaciones han demostrando cómo concentraciones elevadas de reactantes de fase aguda son capaces de predecir la aparición de intolerancia a los hidratos de carbono y/o DM2. Los datos relacionando fenómenos inflamatorios con el síndrome de resistencia a la insulina inicialmente aparecieron en forma experimental in vitro, y luego han sido reproducidos consistentemente en humanos (7).
Uno de los programas investigativos más representativos en este sentido fue realizado en el Estudio de Resistencia a la Insulina y Aterosclerosis (IRAS del inglés Insulin Resistance Atherosclerosis Study), por investigadores de la Universidad de Texas en San Antonio, en cooperación con las Universidades de Vermont (Burlington) y la Escuela Universitaria de Medicina de Wake Forest, en Carolina del Norte. Este estudio incluyó 1.088 pacientes sin enfermedad coronaria, con edades comprendidas entre 40 y 69 años, y evaluó la relación entre marcadores de resistencia a la insulina e indicadores de inflamación: PCR, recuento de leucocitos y fibrinógeno. De ellos 33% tenían intolerancia a los hidratos de carbono al ser incluidos. Utilizando la prueba intravenosa de tolerancia a la glucosa con muestreo frecuente (FSIGTT) para determinar el índice de sensibilidad a la insulina, al igual que inmunoensayos ultrasensibles para medir PCR, los investigadores encontraron una correlación positiva entre los niveles de los tres marcadores de inflamación y las mediciones del contenido total corporal de grasa, concentraciones de insulina y de proinsulina, y una correlación negativa con el índice de sensibilidad a la insulina. Los datos del estudio IRAS muestra a nivel epidemiológico cómo la inflamación crónica es parte del síndrome de resistencia a la insulina (7).
Por otro lado, la inflamación relacionada con resistencia a la insulina induce disfunción endotelial. Estudios recientes sugieren que la insulinorresistencia y la inflamación podrían localizarse en el endotelio, de tal manera que en arterias de gran calibre se produciría aterogénesis, mientras que en la microvasculatura la disfunción endotelial tendría como consecuencia DM2. Esto último debido al estrecho y amplio contacto que existe entre el endotelio microvascular y tejidos sensibles a la insulina, tales como el tejido adiposo o el músculo esquelético (8).
Un grupo de investigadores de la Escuela Médica de Harvard, en Boston, Massachussets, analizó la relación entre la disfunción endotelial y el riesgo de desarrollar DM2 (9). Con base en los datos obtenidos a partir del Estudio de las Enfermeras, de diseño prospectivo de casos y controles, iniciado en 1976 y que enroló 121.700 mujeres estadounidenses cuyas edades oscilaban entre 30 y 55 años, el doctor James Meigs y su grupo de colaboradores analizaron la relación entre los niveles de marcadores de disfunción endotelial (E-selectina, molécula de adhesión intercelular 1 (ICAM-1), molécula de adhesión de células vasculares 1 (VCAM-1)), y el riesgo de desarrollar DM2 (9). Las mujeres incluidas en el estudio fueron sometidas cada dos años a cuestionarios específicamente dirigidos a establecer su estado general de salud.
Durante el período comprendido entre 1989 y 1990, no se reportó aparición de DM2, enfermedad coronaria, cáncer o enfermedad cerebrovascular (ECV) en 32.826 participantes. De ellas, hacia el año 2000, 737 mujeres desarrollaron DM2. Para este grupo se tomaron controles, apareados con base en su raza, edad al momento del diagnóstico de DM2 y niveles basales de glicemia. Además, para las mujeres con mayor índice de masa corporal (IMC), los investigadores utilizaron un grupo control distinto, con el fin de eliminar la obesidad como factor de confusión en los análisis posteriores. Luego de estas maniobras, el grupo control final fue de 785 personas.
El diagnóstico de DM2, al igual que en múltiples estudios epidemiológicos, fue establecido con base en la información suministrada por las pacientes, y confirmada mediante un formulario suplementario, el que incluía averiguaciones acerca de tratamientos hipoglicemiantes, y presencia de síntomas clásicos de diabetes mellitus. Igualmente se tomaron datos de glicemia, primero con base en los criterios para diagnóstico de diabetes emitidos por la Organización Mundial de la Salud hasta 1998. Luego de esta fecha se adoptaron los criterios de la Asociación Americana de Diabetes (en al menos dos mediciones: glicemia al azar > 200 mg/dL, o glicemia basal > 126 mg/dL o > 200 mg/dL a las 2 horas en la prueba oral de tolerancia a la glucosa) (10). Tanto en el grupo de estudio como en el de control se realizaron determinaciones séricas de E-selectina, ICAM-1 y VCAM-1, tomando como resultado principal el riesgo de aparición de DM2.
Luego de un seguimiento de 10 años, niveles elevados de los marcadores de disfunción endotelial mencionados incrementaron en forma significativa el riesgo de desarrollar DM2 en todas las pacientes. Después de ajustar los datos de acuerdo con factores de riesgo cardiovasculares, tales como el índice de masa corporal IMC (y circunferencia de la cintura), antecedentes familiares de DM2, tabaquismo, hábitos alimentarios, consumo de alcohol, actividad física y presencia o no de menopausia, los niveles de E-selectina y de ICAM-1 permanecieron como factores predictores significativos de DM2. Los riesgos relativos ajustados fueron respectivamente 5.08 (95% CI, 3.05 - 8.47) y 2.46 (95% CI, 1.50 - 4.03). Los de VCAM-1 perdieron su poder en los análisis ajustados (RR: 1.05 CI 95%, 0.66-1.70). ICAM-1 y E-selectina conservaron significancia estadística, en forma independiente de la presencia previa de DM2, enfermedad cardiovascular preexistente, hipertensión arterial, dislipidemia, y concentraciones de PCR. Aún más, el poder predictor de ICAM-1 y E-selectina fue independiente de los niveles basales de insulina y de hemoglobina glicosilada A1 c al ingreso en el estudio. De manera interesante, las mujeres más obesas y con mayores concentraciones de E-selectina tuvieron 13.6 veces mayor riesgo (P<0.001) de desarrollar DM2 con respecto a las pacientes más delgadas del estudio, quienes a su vez exhibieron los menores niveles del marcador. Los datos con respecto a ICAM-1 mostraron un patrón similar. De acuerdo con estos resultados, los niveles elevados de ICAM-1 y E-selectina elevan el riesgo de DM2 en 1.5 y 7.5 veces respectivamente en mujeres inicialmente no diabéticas, de manera independiente de otros factores de riesgo cardiovascular (9).
La información aportada por el grupo del doctor Meigs permite postular un mecanismo por medio del cual la inflamación podría contribuir en la patogénesis de la resistencia a la insulina, aterogénesis y DM2, a través de disfunción endotelial, mediada por moléculas de adhesión intercelular (9). En respuesta a citoquinas inflamatorias, en particular factor de necrosis tumoral a (TNF-a) e interleuquina 6 (IL-6), aumenta la producción hepática de reactantes de fase aguda (en particular PCR) y, por parte de las células endoteliales, de E-selectina y de ICAM-1, la cual también puede ser producida por células de la línea blanca hematopoyética. Por su parte, las moléculas de adhesión facilitan la migración de células mediadoras de la inflamación hacia el espacio subendotelial, evento precoz en la formación de placas ateroscleróticas (5).
Estas investigaciones están en concordancia con estudios previos realizados en condiciones experimentales según los cuales el daño genético endotelial inducido [mediante ablación de genes responsables de la síntesis de óxido nítrico (NO)] se acompaña de resistencia a la insulina (11); al igual que múltiples estudios epidemiológicos, realizados en individuos con elevado riesgo de diabetes mellitus (12, 13). De manera interesante, en el estudio sobre riesgo de aterosclerosis en comunidades (ARIC), se encontró un incremento de 71% en el riesgo de desarrollar DM2 en quienes presentaban adelgazamiento arteriolar retiniano, utilizado como un marcador simple de disfunción endotelial, con respecto a quienes no lo tenían (14).
Tejido adiposo como mediador inflamatorio y de resistencia a la insulina.
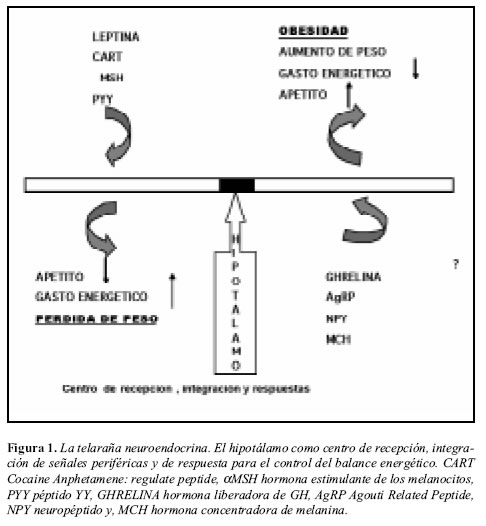
El antiguo concepto del tejido graso como un simple depósito de energía, sin participación metabólica activa ha sido abandonado desde hace más de una década. Un gran cúmulo de evidencia demuestra que el tejido adiposo, incluyendo adipocitos, preadipocitos y tejido estromal, es un órgano metabólicamente activo el cual desempeña un papel central en la homeostasis energética. De hecho, es una estructura capaz de alterar sus propias características funcionales y morfológicas de acuerdo con las condiciones fisiológicas o patológicas predominantes (Figura 1). En la actualidad el tejido adiposo se considera como un órgano endocrino muy importante, y se han identificado múltiples sustancias producidas en él, con actividad tanto paracrina como endocrina (Figura 2) (15). Factores tan importantes como los ácidos grasos libres (AGL) son liberados del adipocito y se encuentran fuertemente relacionados tanto como causa y consecuencia de resistencia a insulina y DM2 (16, 17). Provenientes de la lipólisis que no es adecuadamente suprimida en condiciones de resistencia, los AGL interfieren con la señalización intracelular desencadenada por la insulina y con el metabolismo de los hidratos de carbono (18).También el tejido adiposo es capaz de producir péptidos con influencia sobre las cascadas inflamatorias, procoagulantes, antifibrinolíticas y vasoactivas (19), el término recientemente acuñado de adipocitoquinas o adipoquinas refleja en parte el nuevo concepto acerca del tejido graso, a la vez que sugiere una influencia directa sobre la inflamación.
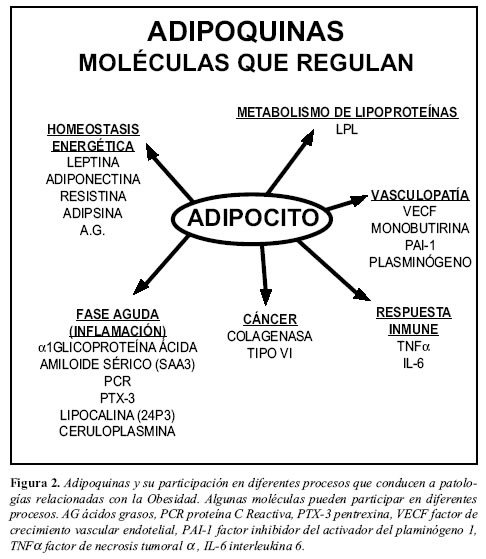
Adipoquinas proinflamatorias
Leptina
Este polipéptido descubierto en 1994 ha demostrado tener un papel central en la homeostasis energética. Es producida por el tejido adiposo, y sus concentraciones son proporcionales a la masa de tejido graso corporal (19). Hasta el momento han sido caracterizados cinco isoformas de receptores de leptina, de las cuales la más estudiada es la Ob-Rb, que a su vez pertenece a la familia de receptores transmembrana para citoquinas. La leptina circula en sangre periférica unida a la forma soluble del receptor (Ob-R).
El mecanismo de acción de la leptina a través del receptor Ob-Rb implica activación de vía de la jak-stat. Esta proteína posee dos sitios principales de acción. En el sistema nervioso central (núcleo arcuato del hipotálamo), regula los mecanismos del apetito, reduciendo el consumo alimentario y aumentando el gasto de energía. Así, la administración de leptina en ratones genéticamente incapaces de sintetizarla (Ob/Ob), reduce la masa adiposa y en consecuencia el peso corporal (20). Inyectada en el sistema nervioso central, la hormona además revierte la hiperglicemia e hiperinsulinemia en ratones Ob/Ob, y mejora la sensibilidad a la insulina, probablemente mediante activación de vías adrenérgicas en el sistema nervioso central (21, 22).
De otro lado, en tejidos periféricos como hígado y músculo esquelético, la leptina mejora también los índices de sensibilidad a la insulina. En este caso se ha propuesto como mecanismo probable la activación de la proteína kinasa activada por 5´AMP (AMPK), esta enzima al inhibir la formación de malonil CoA disminuye el consumo de ATP y la formación de diacil glicerol, implicado en la resistencia a la insulina, mientras que la AMPK estimula vías catabólicas productoras de ATP, en particular el transporte celular de glucosa, al igual que la oxidación de AG y de glucosa (23, 24). El resultado neto de estas acciones sería aumento en la sensibilidad a la insulina y aumento en gasto energético, los cuales favorecen la reducción de tejido adiposo (25).
La leptina ha sido implicada también en el control de la función reproductiva, en la modulación de otros ejes neuroendocrinos, en particular la secreción de hormona de crecimiento (GH), prolactina (PRL). Recientemente además ha sido propuesta la influencia de la hormona sobre la función inmunológica, que además sugiere una relación con el estado nutricional (26). El ayuno prolongado y la ausencia de leptina en ratones (Ob/Ob) se acompañan de inmunosupresión celular (a expensas en especial de células T), la cual puede ser revertida experimentalmente por la administración de leptina, en forma independiente de la recuperación nutricional (27).
Resistina
En el año 2001, durante investigaciones realizadas acerca de los efectos de las tiazolidinedionas (TZD) sobre el tejido adiposo, fue descubierta una sustancia inducible durante la diferenciación adipocitaria, pero susceptible de ser regulada en baja mediante el uso de TZD (28). Los investigadores bautizaron a esta molécula como resistina, creyendo haber encontrado una pieza clave en el rompecabezas de la insulinorresistencia.
Este péptido de 114 aminoácidos es producido en el tejido adiposo blanco durante la diferenciación de los adipocitos, al igual que en adipocitos maduros; sus concentraciones disminuyen (en animales de experimentación) durante el ayuno prolongado, y aumentan con la ingestión alimentaria. La administración de resistina en ratones normales induce resistencia hepática a la insulina y deteriora la captación periférica de glucosa, cambios reversibles al neutralizar el péptido por medios inmunológicos y al administrar TZDs (29). Estos datos, sumados a reportes acerca de la capacidad de la resistina para impedir la inhibición de la producción hepática de glucosa mediada por insulina (30) permitían explicar en parte los efectos de la adiposidad sobre la resistencia a la insulina. Sin embargo, los informes de diversos estudios en animales y en humanos han sido contradictorios.
De hecho, algunos no han logrado demostrar en humanos una relación significativa entre los niveles de resistina, índices de sensibilidad a la insulina o IMC, y han encontrado niveles bajos de RNA mensajero (mRNA) para resistina en tejido adiposo blanco, con respecto a células no adiposas (31, 32). Existen además reportes contradictorios acerca de la expresión diferencial de mRNA para resistina en tejido adiposo subcutáneo, visceral y al interior de los miocitos del tejido esquelético, lo que complica aún más la relación entre este péptido, obesidad y resistencia a la insulina, la que está estrechamente relacionada con el depósito regional visceral e intramiocitario de lípidos (33-35).
Diferencias estructurales en la resistina humana con respecto a aquélla de ratón de 59%, interferencia de otras adipoquinas sobre la secreción de resistina, problemas en la metodología para la medición de la resistina y dudas acerca de la equivalencia que existe entre los niveles séricos y tisulares de resistina son algunas de las explicaciones para reportes tan disímiles en la literatura mundial. Sin embargo, la satisfacción inicial al postular a la resistina como el eslabón perdido en la obesidad y resistencia a la insulina ha sido reemplazada por una razonable incertidumbre acerca del papel de la molécula. Nuevas técnicas de medición del péptido y de su expresión genética, al igual que novedosos modelos experimentales son necesarios antes de concluir al respecto de esta adipoquina.
Factor de necrosis tumoral a (TNF-a)
El TNF-a fue una de las primeras citoquinas identificadas e implicadas en la respuesta inflamatoria sistémica. En pacientes sometidos a estrés agudo es un potente mediador proinflamatorio, y su administración crónica es capaz de inducir caquexia, tal como sucede en pacientes con neoplasias malignas. Sin embargo, TNF-a también ha sido identificado como una adipoquina, y ha sido estrechamente relacionado con el desarrollo de resistencia a la insulina, obesidad y DM2 (36). El TNF-a es una proteína transmembrana de 27 kDa, la cual es clivada para dar origen a una forma soluble de 17 kDa, que es la que posee actividad biológica.
El TNF-a actúa a través de receptores proteicos de membrana (TNFR), de los cuales se han caracterizado dos subtipos: TNFR1 y TNFR2. Al igual que TNF-a, existe una fracción soluble de los TNFR (sTNFR), cuyos niveles son utilizados para caracterizar y cuantificar la actividad del mediador inflamatorio (36), puesto que la citoquina actúa principalmente en forma paracrina.
Los niveles de TNFR y de mRNA para TNF-a se correlacionan positivamente con las cifras de presión arterial, con el peso corporal y con la presencia de DM2 (29, 36). La pérdida de peso reduce los niveles de TNF-a.
La actividad del TNF-a sobre la resistencia a la insulina ha sido explicada mediante varias teorías. Entre otras, se ha propuesto aumento en la liberación de AGL de los adipocitos, interferencia con la síntesis de adiponectina con actividad insulinosensibilizante, e interferencia con la señalización intracelular mediada por receptores de insulina (36). TNF-a, en concentraciones elevadas en el tejido adiposo, interfiere con la actividad de fosforilación de residuos de tirosina en el primer sustrato del receptor de insulina (IRS-1), necesaria para la progresión de la señal intracelular de la hormona. Induce por el contrario fosforilación de residuos de serina en el IRS-1, y desensibilización frente a las acciones de la insulina (37). Además, mediante actividad paracrina, TNF-a activa el factor nuclear kB (NF-kB), lo que produce aumento en la expresión de moléculas de adhesión en la superficie de células endoteliales y células vasculares musculares lisas (CVML) (38). Hay también activación de la población de Th1 de linfocitos T, producción de proteína quimiotáctica de monocitos y macrófagos (MCP-1) y factor estimulante de colonias de monocitos-macrófagos (M-CSF) en macrófagos activados, superóxido dismutasa, e intereleuquinas proinflamatorias (en especial IL-6 e IL-1). El resultado global es la activación y perpetuación de un estado inflamatorio en el tejido adiposo, disfunción endotelial y finalmente, aterogénesis (39, 40).
Si bien existen algunos reportes discordantes en la literatura, el papel del TNF-a como una adipoquina inductora de resistencia a la insulina y en general de un medio inflamatorio en el tejido adiposo parece cada vez más real. Investigaciones en las cuales se demuestra cómo inhibir experimentalmente la actividad del TNF-a mejora la sensibilidad a la insulina apoyan esta hipótesis (40).
Interleuquina 6 (IL-6)
Al contrario del TNF-a, IL-6 se encuentra en grandes concentraciones en el torrente sanguíneo, y ejerce su efecto en forma endocrina. Es producida en múltiples tipos celulares, en particular en el tejido inmunológico, endotelio, muscular y adiposo. Algunos informes sitúan el sitio de producción en el tejido estromal adipocitario (41, 42). Al igual que la leptina, actúa por medio de un receptor proteico transmembrana (gp 130), por medio del cual reduce y perpetua reacciones inflamatorias.
Los niveles circulantes de IL-6 se correlacionan estadísticamente con el IMC, con presencia de resistencia a la insulina y con intolerancia a los hidratos de carbono (17, 43). En algunos estudios la IL-6 predice además el riesgo de desarrollar diabetes mellitus tipo 2 e infarto agudo de miocardio (43, 44). En sujetos sanos la infusión de IL-6 incrementa la glicemia, induce insulinorresistencia y dislipidemia (43). Por su parte, la reducción de peso disminuye su concentración tanto circulante como en adipocitos (29).
No es claro el mecanismo de acción de IL-6 sobre la sensibilidad a la insulina. Sin embargo, parece inducir supresión de señales intracelulares de citoquinas, mediante estímulo de la expresión del supresor de señal de citoquinas 3 (SOCS 3), lo que afecta negativamente la transducción intracelular tanto de insulina como leptina (45). Además, IL-6 antagoniza la secreción de adiponectina (43).
El panorama de IL-6 se complica al evaluar sus efectos en el SNC. Los niveles en SNC de la citoquina exhiben una relación inversa con el contenido adiposo, mientras que la infusión de IL-6 aumenta el gasto energético, sugiriendo en forma paradójica un papel preventivo en la obesidad (46).
Desde hace una década vienen apareciendo reportes acerca de otras moléculas implicadas en inflamación, disfunción endotelial y compromiso del sistema fibrinolítico. Estas sutancias, entre las que resaltan la proteína quimioatrayente de macrófagos y monocitos 1 (MCP-1), el inhibidor del activador del plasminógeno 1 (PAI-1) han demostrado también ser adipoquinas (39). Componentes del sistema renina angiotensina aldosteona (SRAA) como la angiotensina II también son expresados en tejido adiposo, y sus acciones (retención de sodio, aumento del tono y proliferación vasculares) proveen, sumadas a disfunción endotelial, otro eslabón de unión entre la obesidad y la hipertensión arterial (15,17).
Adipoquinas antiinflamatorias
Adiponectina
Desde su caracterización en 1995, esta adipoquina ha sido llamada por nombres tan diversos como adipoQ, apM1, AcrP30 y proteína ligadora de gelatina. Es secretada específicamente en adipocitos como una proteína de 30 kDa, conserva gran homología con el colágeno tipo VIII y X, al igual que con el componente C1q del complemento. Sus niveles se correlacionan negativamente con insulinorresistencia, masa grasa (en particular de distribución visceral), dislipidemia y DM2 (47). En sangre circulan dos isoformas principales de adiponectina, la molécula completa (fAd) y una fracción globular (gAd), resultante del clivaje de fAd. Igualmente han sido clonados dos tipos de receptores de adiponectina, los cuales son proteínas de siete dominios transmembrana, no asociados con proteína G. AdipoR1 es el primero, posee gran afinidad por gAd y es expresado principalmente en tejido muscular esquelético. Por su parte, adipoR2 es más abundante en hígado y su afinidad es intermedia para fAd y gAd (47).
El o los mecanismos por medio de los cuales la adiponectina actúa no han sido aún caracterizados del todo. Sin embargo, uno de ellos parece ser aumentar la oxidación de ácidos grasos en el tejido muscular esquelético, lo cual reduce la cantidad de AGL circulantes inductores de insulinorresistencia (48). Adicionalmente se ha propuesto disminución en la producción hepática de glucosa, inhibición de la expresión de moléculas de adhesión endoteliales (ICAM-1, VCAM-1, E-selectina) y de TNF-a.
Algunos estudios han demostrado también supresión de la migración de macrófagos y de su transformación en células espumosas, estímulo de la síntesis de NO, y reducción de la proliferación de la íntima de vasos sanguíneos lesionados (49). En estudios experimentales in vivo e in vitro la adiponectina ha demostrado impedir la aterogénesis hasta en 30%.
La adiponectina parece actuar a nivel vascular mediante estímulo de la actividad de la AMPK (48). Como ya fue mencionado, al ser activada la enzima desencadena una serie de reacciones catabólicas generadoras de ATP (tales como la oxidación de AG) que tienen un efecto sensibilizante frente a las acciones de la insulina. La actividad de TNF-a también es bloqueada por la adiponectina, que impide la fosforilación y activación del factor nuclear kB. De igual manera la activación de AMPK estimula la síntesis de NO, lo que explicaría los efectos benéficos sobre la función endotelial y su actividad antiaterogénica. Por último, la isoforma globular de la adiponectina inhibe la proliferación celular y la producción de especies reactivas de oxígeno, inducidas por LDL oxidadas durante el proceso de formación de placas ateromatosas (50).
Si bien aún hace falta mayor investigación, la adiponectina se perfila como una adipoquina con importante actividad antiinflamatoria, moduladora del daño endotelial e insulinosensibilizante que probablemente brindará alternativas terapéuticas en paciente obesos y diabéticos tipo 2.
Uniendo resistencia a la insulina y aterogénesis
Proteína CD36
El descubrimiento de la expresión de receptores para insulina en macrófagos ha permitido postular un mecanismo por medio del cual podrían vincularse insulinorrresistencia y desarrollo de placas ateromatosas.
Desde hace algún tiempo se ha descrito cómo la resistencia a la hormona bloquea el mecanismo normal de producción de NO en el endotelio, lo que constituye un mecanismo en la aterogénesis (51). Sin embargo un estudio reciente de la Universidad de Columbia en Nueva York aporta nuevos datos, al implicar la proteína CD36.
El péptido, inicialmente caracterizado como una glicoproteína integral de la membrana plaquetaria (glicoproteína IV), pertenece a la familia de receptores barredores clase B en los macrófagos, y es un elemento clave en el reconocimiento, captación e internalización de moléculas oxidadas de LDL en los macrófagos, durante las etapas iniciales de la formación de placas ateroscleróticas (52, 53). Utilizando modelos de ratones Ob/Ob insulinorresistentes, un equipo de investigadores al mando del doctor Alan R Tall, demostró un aumento en la captación de LDL oxidadas por parte de los macrófagos. El incremento estuvo acompañado por un aumento en los niveles de CD36, causados por reducción en el metabolismo de la proteína. De acuerdo con los análisis, el aumento en los niveles de CD36 se asoció con alteraciones, tanto en el número como en la actividad de tirosina kinasa del receptor de insulina, es decir, con interferencia de la señal insulínica dentro de los macrófagos (54). Mediante administración de altas dosis de insulina, que en condiciones experimentales interfiere con la señal insulínica intracelular, inhibidores de la vía de la fosfatidil inositol 3 kinasa (PI3K), mediadora de la acción intracelular de la hormona) y utilizando ratones con ausencia genética de receptores de insulina, se estableció una relación causal entre un defecto en la señalización intracelular de la insulina y el aumento de CD36. Por su parte, la administración de TZD, conocidas por su actividad insulinosensibilizante disminuyó in vivo la cantidad de CD36 en los macrófagos. En presencia de estas sustancias se redujeron además los niveles de glucosa y de insulina, y aumentó el número de receptores de insulina, indicadores de mejoría de la resistencia. Por último, se obtuvieron resultados similares al utilizar TZD en ratones Ob/Ob genéticamente diseñados para carecer además del receptor para colesterol LDL (ratones LDLR-/-), los cuales exhiben tasas de aterogénesis superiores a los ratones Ob/Ob.
Los datos obtenidos abren una perspectiva interesante en el estudio de los mecanismos de la aterogénesis, al tomar en cuenta la expresión de receptores para insulina en macrófagos, y al demostrar experimentalmente que la resistencia a la insulina puede aumentar postranscripcionalmente los niveles de proteína CD36. Esta a su vez está implicada en el reconocimiento de moléculas oxidadas de LDL, y en su procesamiento posterior para producir células espumosas, evento clave en la aterogénesis (54).
¿Cómo ordenar las piezas del rompecabezas?
Integración de mecanismos y teorías evolutivas
Los descubrimientos recientes según los cuales el tejido adiposo secreta adipoquinas han revelado que el exceso de grasa, en particular a nivel visceral, es capaz de crear un "ambiente inflamatorio", con incremento en especial en las concentraciones de TNF-a, IL-6, PAI-1, leptina, fibrinógeno y componentes del SRAA.
Los datos expuestos a lo largo de esta revisión acercan dos tipos de tejidos aparentemente diferentes: el tejido adiposo y el sistema inmune. Además de la homología estructural entre las adipoquinas y algunos componentes del sistema de defensa, estudios realizados en adipocitos, en células T y en macrófagos han revelado vías proinflamatorias similares en procesos tales como la activación del complemento y la producción de citoquinas. De hecho las células precursoras de adipocitos pueden exhibir en condiciones experimentales una actividad fagocítica similar a la de los macrófagos (55).
Otras investigaciones han demostrado una infiltración del tejido adiposo, particularmente visceral, por parte de macrófagos en obesidad (56). Los mediadores inflamatorios no sólo son producidos por los adipocitos, sino por células del sistema reticuloendotelial y por preadipocitos. La expresión de genes que codifican la síntesis de mediadores inflamatorios se encuentra aumentada en el tejido estromal adipocitario, en el que se encuentran los macrófagos y los preadipocitos. Éstos últimos también sintetizan citoquinas al ser estimulados por TNF-a (57).
Podría postularse en este contexto cómo los cambios en el tamaño del tejido adiposo inducidos por la ganancia de peso generarían secreción adipocitaria de citoquinas (TNF-a), la cual, a su vez induciría síntesis y liberación de factores quimioatrayentes de células del sistema retículo endotelial por parte del tejido estromal adipocitario. El resultado final sería infiltración del tejido graso por parte de macrófagos, y perpetuación de la inflamación, disfunción endotelial y aterogénesis (42).
El tejido adiposo realizaría entonces un papel tanto de causa como de objetivo de un estado inflamatorio crónico de bajo grado (Figura 3) y provee una relación directa con otros componentes del síndrome metabólico. La vía final común es la aterosclerosis, causante de enfermedad vascular generalizada, conduciendo a hipertensión arterial, enfermedad coronaria y enfermedad vascular periférica.
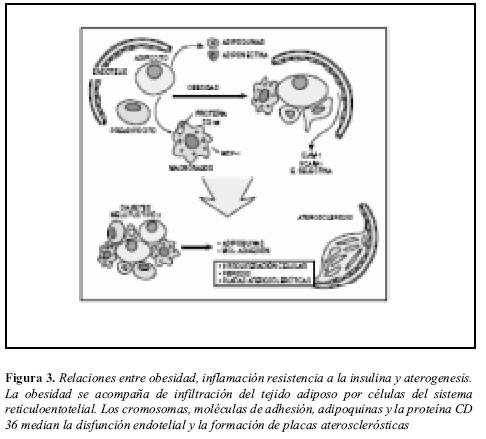
Desde un punto de vista evolutivo, también son evidentes las relaciones entre el sistema inmune y el tejido adiposo, como representante del estado nutricional de los individuos. En insectos como la Drosophila se ha demostrado que las estructuras encargadas de regular el sistema inmune y algunas funciones metabólicas tienen un origen común. En esta especie, en el equivalente del hígado para los mamíferos, se encuentran incluidos el tejido adiposo, el sistema hematopoyético y otros componentes del sistema inmunológico (58). A medida que progresa el desarrollo del animal, cada sistema se especializa progresivamente en la función que le corresponderá cumplir durante su ciclo vital. Este origen común es entendible y ventajoso, puesto que durante el desarrollo de respuestas inmunológicas para controlar infecciones, es imperativo que exista una adecuada reserva energética capaz de sostenerla (42).
La confrontación permanente entre las diferentes especies y las enfermedades infecciosas podría generar, luego de exposición repetida, genotipos "inflamatorios", capaces de inducir la producción de citoquinas proinflamatorias. Se produciría así en algunos individuos un fenotipo "proinflamatorio" crónico de bajo grado que predispondría a la insulinorresistencia (42, 59). Un estudio en tal sentido, realizado por Holgman et al apoya esta teoría, al mostrar que la exposición prenatal a citoquinas puede generar un estado de obesidad e insulinorresistencia después del nacimiento (60).
Puesto que la mayoría de genes implicados en la DM2 son de expresión recesiva, para producir un hipotético "fenotipo inflamatorio" capaz de generar la enfermedad, se requeriría un estado de homozigosidad. Algunas poblaciones han permanecido relativamente aisladas y exhiben un muy bajo grado de mezcla genética, y en consecuencia la posibilidad de aparición del genotipo y fenotipo serían mayores. Tal es el caso de los Indios Pima en Norteamérica, quienes tienen una muy alta prevalencia de obesidad y DM2. Por el contrario, en grupos sometidos a mayor intercambio genético, la posibilidad de homozigosidad sería muy baja, tal como ocurre en la población europea, que exhibe la prevalencia de DM2 más baja del mundo (61).
Evolutivamente uno de los sistemas que más se ha conservado en humanos es el de conservación de energía. De acuerdo con la teoría del Gen Oportunista (Thirfty Gene), en etapas iniciales de nuestra evolución se desarrollaron sistemas altamante efectivos para acumular la escasa energía disponible, dando lugar a la aparición del tejido adiposo. La falta de desarrollo industrial implicaba largas jornadas de ejercicio físico exhaustivo para conseguir cantidades escasa de alimentos. Esta energía debía acumularse de manera eficiente, para ser utilizada posteriormente entre otros, con el fin de defender el organismo frente a innumerables infecciones. El precio, sin embargo, ha sido la susceptibilidad genética, en algunos individuos y grupos poblacionales, a desarrollar un estado inflamatorio crónico en un tejido adiposo cada vez más abundante y disfuncionante. En efecto, la sociedad industrializada ha cambiado las condiciones: ha aumentado dramáticamente el consumo de alimentos ricos en calorías, y se ha reducido de igual manera la actividad física. En Estados Unidos se estima que más del 60% de las personas no participan en programas de actividad física regular, y un 25% sería absolutamente sedentaria (2).
Los sistemas de acumulación energética y de respuesta inmune, por su lado, no parecen haberse adaptado correctamente en todos los casos. Los mecanismos que inicialmente nos permitían sobrevivir podrían haberse vuelto contraproducentes y causantes de inflamación crónica, resistencia la insulina, disfunción endotelial y aterogénesis (Figura 4). En forma alternativa, algunos investigadores postulan que la obesidad podría deberse al desarrollo de resistencia, a nivel del sistema nervioso central, frente a los mecanismos, regulados por insulina y leptina, que controlan la adipogénesis (62).
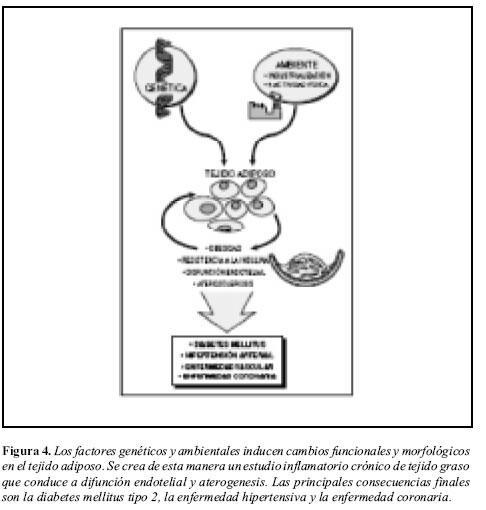
Perspectivas: antiinflamación e insulinorresistencia
En el cada día más amplio espectro del tejido adiposo como órgano endocrino existe un disbalance entre las adipoquinas proinflamatorias y aquellas aparentemente anti-inflamatorias (en especial la adiponectina, cuyo papel ya fue mencionado). Si bien los factores genéticos relacionados con la resistencia a la insulina no son modificables por los métodos disponibles en la actualidad, la carga ambiental sí lo es. Los estudios en los cuales se ha demostrado la participación de mediadores inflamatorios como causa de resistencia se han apoyado también en la influencia que tiene el uso de conductas y sustancias insulinosensibilizantes y antiinflamatorias. Así, la reducción de peso se acompaña de un cambio correspondiente en el tamaño adipocitario, al igual que de disminución en los niveles de adipoquinas pro inflamatorias, mientras que hay aumento en aquellos de adiponectina y mejoría de los índices de disfunción endotelial (63).
La dieta y el ejercicio, en el contexto de la resistencia a la insulina, se comportarían entonces como verdaderos antiinflamatorios.
A nivel farmacológico existen estudios en los cuales el salicilato mejora la sensibilidad a la insulina en modelos de resistencia inducidos por infusión de lípidos, efecto mediado por inactivación de la IkB kinasa-b. La señal insulínica intracelular bajo influencia del antiinflamatorio no es interferida (64), y se han reportado en humanos reducciones significativas de producción hepática de glucosa, glicemia basal y triglicéridos (hasta de 50%), con aumento en la captación y utilización periférica de la glucosa (65).
La aparición de medicamentos insulinosensibilizantes, en particular las TZD, agonistas de los receptores PPAR-g que también se expresan en el tejido adiposo, provee nueva evidencia a favor de la antiinflamación. El uso de TZD reduce los niveles de TNF-a, moléculas de adhesión endoteliales, radicales libres, MCP-1 y PCR, a la vez que aumentan las concentraciones de IL-10 con actividad antiinflamatoria y mejora la sensibilidad a la insulina (66, 67). A nivel clínico, el uso de troglitazona, uno de las primeras TZD utilizadas (ya proscrita) para el manejo de la DM2 evitaba el riesgo de desarrollar la enfermedad en personas de alto riesgo (68), efecto que también se busca esclarecer para las TZD de reciente aparición y para la Metformina.
De igual manera el uso de estatinas, cuyos efectos antiinflamatorios están en boga, más margen en el estudio del Oeste de Escocia (Woscops) una reducción en el riesgo de aparición de DM2 en 30% (69). Probablemente el efecto de la estatinas se relaciona con su capacidad de reducir los niveles de TNF-a, PAI-1 y IL-6 (70) y su probable sinergismo con los PPAR-g (71).
Otro grupo de medicamentos con presunta actividad antiinflamatoria es el de los inhibidores de enzima convertidora de angiotensina (IECA) y el de los bloqueadores de receptores para angiotensina (ARA II). Si bien a nivel experimental los efectos de los últimos no han sido consistentes hasta el momento, en estudios clínicos ambos parecen ejercer una actividad benéfica sobre el riesgo de DM2. Uno de ellos, ramipril, redujo la incidencia de DM2 en 32% con respecto al placebo en el estudio HOPE (72). Por su parte, losartán también se asoció con menor incidencia de la enfermedad, con respecto al atenolol en el estudio LIFE (73). Los mecanismos responsables de estas acciones no son claros actualmente; sin embargo, se ha observado su capacidad de reducir las concentraciones de TNF-a (36).
Últimamente una nueva estrategia terapéutica ha surgido para el síndrome cardiometabólico con la utilización de antagonistas de los canabinoides endógenos. Desde el descubrimiento del principal componente psicoactivo del Cannabis sativa, el D9 tetrahidrocanabinol, grandes avances se han logrado en el estudio del sistema endocanabinoide y su papel en la regulación energética del organismo (74). Los endocanabinoides son derivados del ácido araquidónico, conjugados con etalonamina o glicerol (75). Los canabinoides naturales (D9 tetrahidrocanabinol) y sintéticos se unen a dos tipos de receptores acoplados a proteína G: el receptor de canabinoide tipo 1 (CB1) y el receptor de canabinoide tipo 2 (CB2) (76). El CB1 se encuentra en forma más abundante en el cerebro y se ha relacionado con la actividad orexiante de los canabinoides y con sus acciones sobre el metabolismo lipídico. Por su parte, CB2 se concentra en las células del sistema inmunológico (74). Entre los canabinoides endógenos se han identificado el amida N-araquidonoiletanolamina (anandamide) y el monoacil-glicerol 2-araquidonoilglicerol (2-AG) (76). El anandamide se comporta como agonista parcial de CB1 y CB2, con mayor afinidad por el receptor CB1. El 2-AG se comporta como agonista completo, aunque con menor afinidad por ambos receptores (75).
Los efectos de los endocanabinodes sobre la regulación del apetito a nivel central parecen ocurrir a dos niveles. Se propone que los endocanabinoides estimulan continuamente la búsqueda y consumo de alimentos. La actividad endocanabinoide puede también ser activada en el hipotálamo, luego de un ayuno corto (74).
Sin embargo, los endocanabinoides no parecen limitarse al control del apetito. En el estudio DIO (Diet induced obesity) en ratones, el antagonismo de los receptores CB1 por rimonabant causa una pérdida de peso mayor de la esperada para la actividad anorexiante demostrada (77). Se ha sugerido que el antagonismo con rimonabant podría tener efecto sobre la lipogénesis y la acumulación de grasa (74).
Osei-Hyiaman y colaboradores demostraron que la activación de receptores CB1 causa aumento de la expresión de genes fundamentales para la lipogénesis como el factor de transcripción SREBP-1 (Sterol Responsive Element Binding Protein 1) y sus enzimas blanco (la acetyl CoA carboxilasa 1 y la sintetasa de ácidos grasos). El tratamiento con agonista CB1 aumentó la síntesis de grasa hepática, los niveles de anandamide y de CB1, efectos que son bloqueados por antagonistas de CB1. A nivel hipotalámico los resultados fueron similares. Concluyen los investigadores que el anandamide contribuye a la obesidad, y que la vía de la sintetasa de ácidos grasos puede ser una ruta común para la acción canabinoide central y periférica (78).
Los ensayos clínicos en humanos con rimonabant son alentadores. En el 2004 se conocieron los resultados del estudio RIO-North America. En este estudio de fase III, multicéntrico, doble ciego, 3.040 pacientes con una dieta hipocalórica fueron asignados en forma aleatoria a manejo con placebo o rimonabant (5 o 20 mg/día) por dos años. Luego del primer año de tratamiento los pacientes que inicialmente recibieron rimonabant se reasignaron a placebo o continuación de rimonanabant.
Tras dos años de tratamiento, la circunferencia abdominal se redujo 8 cm con 20 mg de rimonabant, 4.9 cm con dosis de 5 mg y 3.8 cm con placebo (p<0.001). Durante el período de estudio, el 32.8% de los pacientes tratados con 20 mg perdió más de 10% del peso inicial, 20% en el grupo de 5 mg/día y 16.4% en quienes recibieron placebo (p<0.001). El colesterol HDL aumentó un 24.5% durante el periodo de tratamiento con 20 mg/día de rimonabant, mientras que con 5 mg y con placebo se evidenció un aumento de 15.6% y de 13.8% (p<0.001) respectivamente. Los triglicéridos se redujeron en 9.9% en pacientes tratados con rimonabant 20 mg/día y un 1.6% con placebo (p <0.05). Adicionalmente, los pacientes que recibieron rimonabant mejoraron significativamente su sensibilidad a la insulina. En el grupo de pacientes tratados con rimonabant 20 mg por dos años se redujo la incidencia de síndrome metabólico en más de un tercio (p<0.001) (74).
En abril de 2005 se publicó otro ensayo clínico con rimonabant. Esta vez RIO-Europe estudió a 1.507 pacientes con índice de masa corporal (IMC) >30kg/m2 o con IMC >27kg/m2 e hipertensión y/o dislipidemia. El seguimiento se realizó a un año.
La pérdida de peso fue significativamente mayor para los pacientes tratados con rimonabant (ambas dosis) que para los pacientes tratados con placebo. El grupo de rimonabant 20 mg mostró mejoría significativa en la circunferencia de cintura, colesterol HDL, triglicéridos, resistencia a la insulina y aparición de síndrome metabólico (79).
La evidencia experimental y clínica acumulada sobre endocanabinoides es alentadora, y aunque todavía falta conocer mejor sus efectos sobre los sistemas reguladores del apetito a nivel central y sobre el balance energético periférico, lo aprendido hasta ahora abre interesantes caminos que en corto tiempo ya se han traducido en nuevas posibilidades terapéuticas para el síndrome cardiometabólico.
El efecto que tienen diversas estrategias antiinflamatorias y moduladoras de la función endotelial ha sido utilizado como una prueba de la relación existente entre inflamación, resistencia a la insulina y aterogénesis. Sin embargo, todos los datos abren una interesante perspectiva terapéutica hacia el futuro, al mostrar las ventajas de la antiinflamación.
A pesar de la verdadera explosión de investigaciones que vienen apareciendo en la última década, muchas preguntas permanecen aún sin responder, relacionadas con los mecanismos por medio de los cuales se dispara una cascada inflamatoria en el tejido adiposo y cómo esto induce finalmente la resistencia a la insulina. No conocemos todavía a profundidad las intimidades de la interlocución molecular entre los receptores de insulina y la inflamación ni cuál es el estímulo inicial para la infiltración específica del tejido adiposo por parte de células del sistema reticuloendotelial. De hecho no sabemos aún si los macrófagos son atraídos por una reacción inflamatoria inducida por los adipocitos disfuncionantes, o si éstos infiltran inicialmente la grasa y luego desencadenan inflamación. Es todavía en gran medida desconocido el papel relativo de cada una de las células (vasculares, preadipocitos, adipocitos, macrófagos) en la cascada fisiopatológica. Sin duda los próximos años serán claves para cimentar los conocimientos que emergen en estos momentos, piezas del cubo de Rubik.
Por último, en la actualidad se utilizan como marcadores de riesgo cardiovascular y de homeostasis metabólica marcadores muy poco precisos, tales como la glicemia o los niveles de lípidos o las cifras de presión arterial. Si bien son invaluables, no reflejan, a la luz de los conocimientos actuales, la telaraña de alteraciones metabólicas que finalmente conducen hacia la aterogénesis. Mayor atención deberá entonces prestarse a los factores de riesgo no tradicionales, y comenzar a utilizar otros indicadores, tales como el fenotipo de las lipoproteínas de baja densidad, apoproteína B, PCR, PAI-1 fibrinógeno y marcadores de disfunción endotelial, entre otros. El desarrollo de nuevos modelos experimentales y clínicos diseñados específicamente para "disecar" toda la interrelación entre el sistema inmunológico y el sistema de acumulación de energía permitirá en unos años completar la mayoría de los elementos de un rompecabezas en el que cada día aparecen más piezas.
Referencias
1. King H, Aubert RE, Herman WH. Global Burden of diabetes, 1995- 2025:Prevalence, numerical estimates and projections. Diabetes Care. 1998; 21:1414-31 [ Links ]
2. Stein CJ, Graham AC. The Epidemic of Obesity. J Clin Endocrinol Metab 2004; 89:2522-5 [ Links ]
3. Stratton IM, Adler AI, Holman RR, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS): prospective observational study. BMJ. 2000; 321: 405-12. [ Links ]
4. Lastra G, Duarte DE:Sensibilidad a la insulina en Hipertensos no diabéticos tratados con Bloqueadores de los Canales de Calcio Acta Med Colomb 1998;23:288-95. [ Links ]
5. Libby P. Changing Concepts in Atherogenesis. J Inter Med 2000; 247: 349-58. [ Links ]
6. Pinkney JH, Stehower CD, Coppak SW. Endothelial Disfunction: cause if the Insulin Resistance Syndrome. Diabetes 1997; 46 (Suppl 2):S9-13 [ Links ]
7. Festa A, D'Agostino Jr R, Howard G, Haffner SM. Chronic Subclinical Inflammation as Part of the Insulin Resistance Syndrome The Insulin Resistance Atherosclerosis Study (IRAS). Circulation 2000;102:42-7. [ Links ]
8. Clark MG, Wallis MG, Barrett EG. Blood flow and muscle metabolism: a focus on insulin action. Am J Physiol Endocrinol Metab 2003; 284: E241-58. [ Links ]
9. Meigs JB, Hu FB, Rifai N. Biomarkers of Endothelial Dysfunction and Risk of Type 2 Diabetes Mellitus. JAMA 2004; 291: 1978-86. [ Links ]
10. American Diabetes Association: Report of the Expert Comittee on Diagnosis and Classification of Diabetes Mellitus. Diabetes 1997;20:1183-97. [ Links ]
11. Duplain H, Burcelin R, Sartori C, et al. Insulin Resistance, hyperlipidemia and impaired hemostasis in mice lacking endothelial nitric oxide synthase. Circulation 2001;104:342-5. [ Links ]
12. Caballero AE, Arora S, Saouaf R, et al. Microvascular and macrovascular reactivity is reduced in subjects at risk for type 2 diabetes mellitus. Diabetes 1999; 48:1856-62. [ Links ]
13. Vehkavaara S, Seppala-Lindroos A, Westerbacka J, et al: In vivo endothelial dysfunction characterizes patiens with impaired fasting glucose. Diabetes Care 1999; 22: 2055-60. [ Links ]
14. Duncan BB, Schmidt MI, Offenbacher S, et al: Factor VIII and other hemostasis variables are related to incident diabetes in adults: The Atherosclerosis Risk in Communities (ARIC) Study. Diabetes Care 2003; 26:1745-51. [ Links ]
15. Ahima RS, Flier JS. Adipose tissue as an endocrine organ. Trends Endocrinol Metab 2000; 11:327-32. [ Links ]
16. Bays H, Mandarino L, De Fronzo R. Role of the adipocyte, freee fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferators-activated receptor agonists provide a rationale therapeutic approach. J Clin Endocrinol Metab 2004; 89:463-78. [ Links ]
17. Kershaw EE, Flier JS, Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004; 89:2548-56. [ Links ]
18. Lyon, CJ, Law RE, Hsueh WA. Minireview: adiposity, inflammation and atherogenesis. Endocrinology 2003; 144:2195-200. [ Links ]
19. Considine RV, Sinha MK, Heiman ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 1996; 334:292-5. [ Links ]
20. Halaas JL, Gajiwala KS, Maffei M, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995; 296:543-6. [ Links ]
21. Ahima RS, Prabakaran D, Matanzoros C. Role of leptin in neuroendocrine response to fasting. Nature 1996; 382: 250-2. [ Links ]
22. Pelleymounter MA, Cullen MJ, Baker MB. Effects of the obese gene product on body weight regulation in Ob/Ob mice. Science 1995:269: 540-3. [ Links ]
23. Minokoshi Y, Kim YB, Peroni OD, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002; 415: 339-43. [ Links ]
24. Minokoshi, Y, Kahn BB. Role of AMP-activated protein kinase in leptin- induced fatty acid oxidation in muscle. Biochem Soc Trans 2003; 31: 196-201. [ Links ]
25. Rajala MW, Scherer PE. Minireview: The adipocyte:-At the crossroads of energy homeostasis, inflammation and atherosclerosis. Endocrinology 2003; 144:3675-773. [ Links ]
26. Huang L, Li C. Leptin: a multifunctional hormone. Cell Res 2000; 10: 81-92. [ Links ]
27. Lord GM, Matarese G, Howard JK, et al. Leptin modulates the T cell immune response and reverses starvation-induced immunosupression. Nature 1998; 394: 897-901. [ Links ]
28. Steppan CM, Bailey ST, Bhat S, et al. The hormone resistin links obesity to diabetes. Nature 2001; 409:307-12. [ Links ]
29-Pittas AG, Joseph NA, Greenberg AS. Hot topic: Adipocitokines and Insulin Resistance. J Clin Endocrinol Metab 2004; 89: 447-52. [ Links ]
30. Rajala MW, Obici S, Scherer PE, Rosetti L. Adipose-derived resistin and gut-derived esistin resistin-like molecule selectively impair insulin action on glucose production. J Clin Invest 2003; 111:225-30. [ Links ]
31. Janke E, Gorzelniak K, Luft FC. Resistin gene expression in adipocytes is not related to Insulin resistance. Obesity Research 2002; 10: 1-5. [ Links ]
32. Vidal-Puig A, O'Rahilly S. Resistin: a new link between obesity and Insulin Resistance?. Clin Endocrinol (Oxford) 2001; 55: 437-8. [ Links ]
33. Kelley DE, Goodpaster BH, Skeletal Muscle Triglyceride: An aspect of regional adiposity and insulin resistance. Diabetes Care 2001; 24: 933-41. [ Links ]
34. Savage Db, Sewter CP, Klenk ES, et al: Resistin/Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-g in humans. Diabetes 2001; 50: 2199-202. [ Links ]
35. Way JM, Gorgun CZ, Tong Q, et al. Adipose tissue resistin expression is severily upressed in obesity and stimulated by peroxisome proliferator- activated receptor-g gonists. J Biol Chem 2001; 276: 25651-3. [ Links ]
36. Hotamisligil GS, Spiegelman BM, Tumor necrosis factor a: a key component of the obesity-diabetes link. Diabetes 1994; 43: 1271-8. [ Links ]
37. Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of TNF-a. J Clin Invest 1994; 94: 1543-9. [ Links ]
38. Landry DB, Couper LL, Lindner V. Activation of the NF-kB and Ikk system in smooth muscle cells after rat arterial injury. Induction of vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1. Am J Pathol 1997; 151:1085-95. [ Links ]
39. Frostegard J, Ulfgren AK, Nyber P, et al. Cytokine expresión in advanced human atherosclerotic plaques: dominance of proinflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis 1999; 145: 33-43. [ Links ]
40. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor a: direct role in obesity-linked insulin resistance. Science 1993; 259: 87-91. [ Links ]
41-Freid SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissue of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J clin Endocrinol Metab 1998; 83: 847-50. [ Links ]
42. Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 2003; 112: 1785-8. [ Links ]
43. Fernández-Real JM, Ricart W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocrine Reviews 2003; 24: 278-301. [ Links ]
44. Ridker PM, Rifai N, Stampfer MJ, Hennekens, CH. Plasma concentration of interleukin 6 and the risk of future muocardial infarction among apparently healthy men. Circulation 2000; 101: 1767-72. [ Links ]
45. Ueki K, Kondo T, Kahn R. Suppressor of Cytokine Signaling 1 (SOCS-1) and SOCS-3 cause Insulin Resistance though Inhibition of Tyrosine Phosphorylation of Insulin Receptor Substrate Proteins by Discrete Mechanisms. Mol Cell Biol 2004;24:5434-46. [ Links ]
46. Wallenius V, Wallenius K, Ahren B, Rudling M, et al. Interleukin-6- defficient mice develop mature onset obesity. Nature Medicine 2002; 8: 75-9. [ Links ]
47. Goldstein BJ, Scalia R. Adiponectin: A novel adipokine linking adipocytes and vascular function. J Clin Endocrinol Metab, 2004; 89: 2563-2568 [ Links ]
48. Boden G, Shulman GI. Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and b-cell dysfunction. Eur J Clin Invest, 2002; 32 (3): 14-23 [ Links ]
49. Ouchi N, Ohishi M, Kihara S, et al. Association of hypoadinectinemia with impaired vasoreactivity. J. Hipert, 2003; 42: 231-234 [ Links ]
50. Motoshima H, Wu X, Mahadev K, Goldstein BJ. Adiponectin supresses proliferation and superoxide generation and enhances eNOS activity in endothelial cells treated with oxidized LDL. Biochem Biophys Res Commun, 2004; 315: 264-271) [ Links ]
51. Vincent D, Ilany J, Kondo T,et al: The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J Clin Invest, 2003; 111: 1373-1380 [ Links ]
52. Febbraio M, Hajjar DP, Silverstein RL. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation and lipid metabolism. J Clinl Invest, 2001; 108:785-791 [ Links ]
53. Febbraio M, Dondrez EA,Smith JD et al: Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest, 2000; 105: 1049-1056 [ Links ]
54. Chien-Ping L, Seongah H, Okamoto H, et al. Increased CD 36 protein as a response to defective insulin signaling in macrophages. J Clin Invest, 2004; 113: 764-773 [ Links ]
55. Charrière G,Cousin B,Arnaud E, et al: Preadipocyte conversión to macrophage. Evidence of plasticity. J. Biol. Chem, 2003; 278: 9850-9855 [ Links ]
56. Weisberg SP, McCann Ddesai M, et al: Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest, 2003; 112: 1796-1808 [ Links ]
57. Xu H, Barnes GT, Yang Q, et al: Chronic inflammation in fat plays a crucial role in the development of obesity related insulin resistance. J Clin Invest, 2003; 112: 1821-1830 [ Links ]
58. Tong Q, Dalgin G, Xu H et al: Function of GATA transcription factors in preadipocyte- adipocyte transition. Science, 2000; 290: 134-138 [ Links ]
59. Wilson AG, Symmons JA, Mc Dowell TL,et al: Effects of a polymorphism in the human Tumor Necrosis Factor-_ promoter on transcriptional activation. Proc Nat Acad Sc USA, 1997; 94: 3195-3199 [ Links ]
60. Dahlgren J, Nilsson C, Jennishe E, et al: Prenatal cytokine exposure results in obesity and gender-specific programming. Am J Physiol, Endocrinol Metab, 2001; 281: E326- E334 [ Links ]
61. Lev-Ran A. Thirfty genotype: how applicable is it to obesity and type 2 diabetes mellitus? Diabetes Reviews, 1999; 7:1-22 [ Links ]
62. Schwartz MW, Niswender KD. Adiposity Signaling and Biological Defense Against Weight Gain: Absence of Protection or Central Hormone Resistance? J Clin Endocrinol Metab, 2004; 89:5889-5897 [ Links ]
63. Clarkson P, Montgomery HE, Mullen MJ, et al. Exercise training enhances endothelial function in young men. Am J Cardiol, 1999; 33: 1379-1385 [ Links ]
64. Kim J, Kim Y-J, Filmore J, et al. Prevention of fatinduced insulin resistance by salicylate. J Clin Invest, 2001; 108: 437-446 [ Links ]
65. Hundal RS, Petersen KF, Mayerson AB, et al. Mechanism by which high- dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest, 2002; 109: 1321-1326 [ Links ]
66. Ghanim H, Garg R, Aljada A, et al. Supression of Nuclear Factor kB by troglitazone: evidence of an anti-inflammatory effect and a posible antiatheroscletotic effect in the obese. J Clin Endocrinol Metab, 2001; 86: 1306-1312 [ Links ]
67. Haffner SM, Greenberg AS, Weston WM, et al. Effect of rosiglitazone treatment on nontraditional markers of cardiovascular disease in patients with type 2 diabetes mellitus. Circulation, 2002; 106: 679-684 [ Links ]
68. Buchanan TA, Xiang AH, Peters RK, et al. Preservation of pancreatic b-cell function and prevention of type 2 diabetes mellitus by pharmacological treatment of insulin resistance in high-risk hispanic women. Diabetes, 2002; 51: 2796-2803 [ Links ]
69. Freeman DJ, Norrie J, Sattar N, et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of 65.-Scotland Coronary Prevention Study. Circulation, 2001; 103: 357-362 [ Links ]
70. Rosenson RS, Tangney CC, Casey LC. Inhibition of proinflammatory cytokine production by pravastatin. Lancet, 1999; 353: 983-984 [ Links ]
71. Inoue I, Itoh F, Aoyagi S, et al. Fibrate and statin synergistically increase the transcriptional activities of PPAR-g/RXR and increase the transactivation of NFkB. Biochem Biophys Res Commun, 2002; 290:131-139 [ Links ]
72. Yusuf S, Gerstein H, Hoogwerf B,et al: Ramipril and the development of diabetes. JAMA 2001; 286: 1882-1885 [ Links ]
73. Lindholm LH, Visen H, Borch-Johnsen K, et al. Risk of new-onset diabetes in the losartan intervention for endpoint reduction in hypertensión study. J Hyper, 2002; 20: 1879-1886 [ Links ]
74. Di Marzo V, Matias I. Endocannabinoid control of food intake and energy balance. Nature Neuroscience 2005; 8: 585-589 [ Links ]
75. De Fonseca FR, Del Arco I, Bermudez-Silva FJ et al. The endocannabinoid system: physiology and pharmacology. Alcohol and Alcoholism. 2005; 40: 2-14 [ Links ]
76. Lichtman AH, Cravatt BF. Food for thought: endocanabinoid modulation on lipogenesis. J Clin Invest 2005; 115: 1130-1133 [ Links ]
77. Ravinet Trillou C, et al. 2003. Anti-obesity effect of SR141716, a CB1 receptor antagonist, in dietinduced obese mice. Am J Physiol Regul Integr Comp Physiol 2003; 284: R345-R353 [ Links ]
78. Osei-Hyiaman D et al. Endocannabinoid activation at hepatic CB1 stimulates fatty acid síntesis and contributes to diet induced obesity. J Clin Invest 2005; 115: 1298-1305. [ Links ]
79. Van Gaal LF, Rissanen AM, Scheen AJ, Ziegler O, Rossner S. RIO-Europe Study Group. Effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet 2005; 365: 1389-1397. [ Links ]

