










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.30 no.3 Bogotá July/Sept. 2005
Lic. Claudia Tamar Silva: Biol., MSc. Profesor Asistente;
Lic. Dora Janeth Fonseca: Biol., MSc. Profesor Auxiliar;
Dra. Heidi Eliana Mateus: MSc, Profesor Auxiliar,
Lic. Nora Constanza Contreras: Biol., MSc, Instructor Asistente;
Dr. Carlos Martín Restrepo: MSc, PhD(c). Jefe. Unidad de Genética, Instituto de Ciencias Básicas, Facultad de Medicina Universidad del Rosario. Bogotá, D.C.
Conflicto de intereses: Los autores declaran que no tienen intereses de ningún tipo con las empresas comerciales que puedan beneficiarse de la presente investigación.
Correspondencia: Claudia Silva, Dirección: Calle 63D N 24-31, Teléfono: 3474570/60 ext 266, E mail: ctsilva@urosario.edu.co
Recibido: 17/02/05 Aprobado: 13/06/05
Resumen
La distrofia muscular de Duchenne y Becker es la miopatía más común en ni ños y es causada por la ausencia de la proteína distrofina. Los afectados presentan signos de la enfermedad a edades tempranas de la vida, pierden la habilidad para caminar al comienzo de la segunda década y usualmente, fallecen alrededor de los 20 años de edad. El islamiento del gen defectuoso ha llevado a un mejor entendimiento del proceso de la enfermedad y ha permitido el diagnóstico preciso en los afectados, la posibilidad de asesoramiento genético y diagnóstico prenatal, así como la aplicación de nuevas terapéuticas basadas en el conocimiento de la patogénesis de la enfermedad. El propósito de esta revisión es presentar el progreso hecho en estas áreas, refiriéndonos particularmente a la fisiopatología y al diagnóstico molecular de la enfermedad en Colombia.
Palabras clave: Duchenne, Becker, distrofias musculares, distrofina, diagnóstico.
Abstract
Duchenne and Becker's muscular dystrophy is the most common form of muscle dystrophy found in children, and is caused by an absence of the protein dystrophin. Affected boys show signs of the disease early in life, stop walking at the beginning of the second decade, and usually die by age 20. The isolation of the defective gene has led to a better understanding of the disease process and has allowed an accurate diagnosis of affected patients. Moreover, genetic counseling and pre-natal diagnosis, together with the application of new therapies based on the knowledge of the disease's pathogenesis, are now a possibility. The purpose of this review is to present the progress made in this area, emphasizing, in particular, the pathophysiology and the molecular diagnosis of the disease in Colombia.
Key words: Duchenne, Becker, muscular dystrophy, dystrophin diagnosis.
Introducción
La distrofia muscular de duchenne (DMD), también conocida como enfermedad de Meryon, y la forma alélica menos severa, la distrofia muscular de Becker (DMB), son las distrofias más comunes, de herencia recesiva ligada al X, afectan a uno de cada 3.500 niños (1). Tanto la DMD como la DMB presentan gran heterogeneidad de mutaciones en el gen de la distrofina, localizado en el brazo corto del cromosoma X, en la banda Xp21 (2, 3) (Figura 1). El gen consta de 2.500 kb y está conformado por 79 exones, dando como resultado la transcripción de ARNm de aproximadamente 14 kb, el cual codifica para la distrofina, una proteína de 427 kD (4, 5). Este gen tiene una variedad de tipos de empalme y un gran número de promotores, algunos de ellos tejido-específicos, los cuales generan diferentes proteínas en cuanto a longitud y/o secuencia en la región amino-terminal. Se ha demostrado que un promotor localizado en el intron 29, constituye una isoforma de Distrofina de 260 KD, la cual se expresa en la retina y las mutaciones se asocian a ceguera nocturna estacionaria congénita (6).
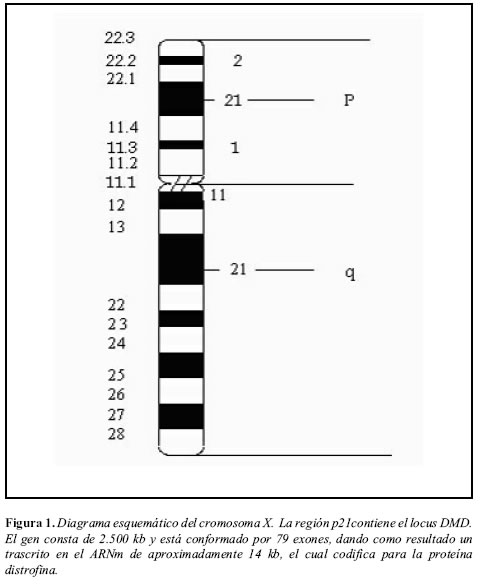
Características clínicas
Los pacientes con DMD y DMB se caracterizan por debilidad y atrofia muscular proximal y luego distal que son progresivas, inestabilidad y dificultad para caminar, caídas frecuentes, pérdida de la habilidad para caminar e imposibilidad de mantenerse erguido, generalmente después de los 12 años (7, 8).
La DMD conduce a la muerte en la segunda década de la vida, ocasionada usualmente por problemas respiratorios o cardíacos, mientras que la DMB permite una sobrevida mejor que puede alcanzar hasta cinco o seis décadas (1). El resumen de los hallazgos principales en estos pacientes según la época de aparición se presenta en la Tabla 1.
Diagnóstico
Aspectos clínicos y paraclínicos
En el laboratorio, una de las alteraciones más características es la elevación (desde el nacimiento) del nivel de fosfocreatin kinasa (CPK) sérica, que puede alcanzar cifras considerables (10 a 50 veces por encima de lo normal). Hay valores elevados de CPK entre los 14 y 22 meses de edad que luego tienden a disminuir, pero siempre se conservan por encima de los valores normales. Con menor importancia, se puede contar con los niveles séricos elevados de aldolasa sérica y deshidrogenasa láctica (LDH).
La electromiografía (EMG) muestra un trazado de tipo miopático no específico, en el cual se observa disminución de la duración media de los potenciales de la unidad motora y un aumento de las formas polifásicas, que reflejan la pérdida de la fibra muscular. Sin embargo, en la EMG convencional pueden obtenerse hallazgos de tipo neurógeno como consecuencia de los mecanismos de denervación y reinervación involucrados en la misma enfermedad (9). Desde 1957, Buchthal describió una técnica original de análisis cuantitativo de potenciales de acción de la unidad motora, que permiten estudios más específicos (10). Otros autores han desarrollado un método que se basa en la medición de "giros" medidos en un patrón de interferencia registrado con una fuerza constante de contracción, encontrando que el número de giros aumenta en los pacientes con miopatía (11); adicionalmente, el número de giros y el promedio de su amplitud se han convertido en un criterio muy sensible para la diferenciación de las miopatías y las neuropatías (9). Cabe resaltar que los hallazgos electrofisiológicos pueden ser diferentes según la etapa de la enfermedad o por la reorganización de las fibras musculares (9).
Por otra parte, el electrocardiograma (ECG) da signos importantes de apoyo para el diagnóstico, ya que en algunos casos se evidencia un claro compromiso del músculo cardiaco.
La biopsia muscular puede llegar a ser necesaria para el diagnóstico. En ausencia de un historial típico, se encuentran generalmente los hallazgos observados en otros tipos de distrofias musculares como:
· Necrosis segmentaria, evidenciada principalmente por la presencia de fagocitos, cambios histológicos variables en las miofibrillas, evidencia de sobrecontracción de las fibras musculares (la longitud del sarcómero es varias veces mayor que su tamaño fisiológico normal) y daño en la membrana (éstos últimos observables por medio de microscopía electrónica).
· Regeneración después de los episodios de necrosis. El proceso de necrosis-regeneración vuelve y comienza, hasta que las células pierden su capacidad reproductiva. Las alteraciones observadas en la fibra muscular pueden presentarse desde edades tempranas, aún antes de que la enfermedad llegue a ser evidente clínicamente (12).
Dentro del estudio de las fibras musculares, además de la biopsia muscular, existe la inmunohistoquímica. En este proceso se utilizan anticuerpos antidistrofina o contra alguno de los componentes del llamado complejo DGC (complejo distrofina-glicoproteínas), evaluándose tanto la cantidad como la calidad de la distrofina y/o de las glicoproteínas asociadas a ella. La ausencia completa de la distrofina o cifras de menos de 3% son específicas y características del fenotipo grave de DMD. En 85% de los pacientes con DMB, la distrofina tiene un peso molecular anormal, al ser más pequeña por deleción (80%), o más grande por duplicación (5%). En 15% de los pacientes restantes, la proteína tiene un tamaño normal (13). Estos hallazgos inmunohistoquímicos se correlacionan generalmente muy bien con el fenotipo e incluso llegan a ser útiles en la determinación del estado de portadora.
La distrofina
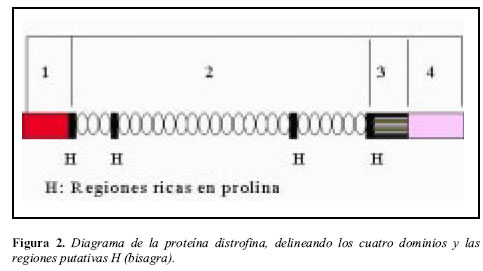
La distrofina es una proteína de 3,685 aminoácidos con cuatro dominios (Figura 2). El primero muestra homología con las regiones de unión al extremo aminoterminal de la a-actinina y de la b-espectrina. El segundo dominio consta de una serie de 24 repeticiones de 109 aminoácidos, las cuales forman una estructura helicoidal triple; estas repeticiones están interrumpidas por regiones ricas en prolina que le añaden flexibilidad a la molécula, actuando como bisagras moleculares. El tercer dominio, es similar a la región de unión al calcio de la a-actinina. El último dominio, consta de 400 aminoácidos y tiene por función formar un complejo con las glicoproteínas de membrana (4, 15).
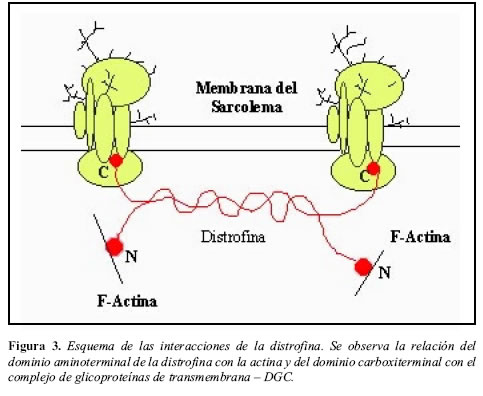
La distrofina se expresa en el sarcolema en el músculo estriado esquelético, músculo liso y estriado cardíaco; también, se encuentra en algunos tipos específicos de neuronas, incluyendo las células de Purkinje y las neuronas de la corteza del cerebro. Aunque la función precisa aún no ha sido establecida, al parecer, el papel de la distrofina es estabilizar las membranas plasmáticas durante la contracción muscular (6,16-18); a través de la unión del dominio aminoterminal a la actina, mientras que el extremo carboxi-terminal se una a las proteínas DGC y éstas, a su vez, se unirían a la laminina en el exterior de la membrana del sarcolema (4,15, 19, 20) (Figura 3).
Mutaciones
Se ha descrito una gran heterogeneidad en las mutaciones del gen de la distrofina que incluyen deleciones, duplicaciones y mutaciones puntuales (21). Las deleciones suman el 70% de todos los casos y afectan a uno o varios exones (22). En Colombia se ha descrito que el 31% de los pacientes tienen deleciones detectables (23, 24), hallazgo que es acorde con lo descrito para otras poblaciones en Latinoamérica (25, 26). Este hallazgo se explica probablemente por la heterogeneidad de nuestro acervo genético, el cual también ha sido observado en otras enfermedades como fibrosis quística (27). Las deleciones se concentran en dos regiones del gen, que son puntos calientes o "hot spots": la mayoría (80%) en los exones 44 al 52 y, dentro de esta región, el 40% se ubican sobre el exón 44, uno de los más extensos del gen de la distrofina (28), el otro punto caliente o "hot spot" se encuentra en la región 5´ terminal del gen y comprende los exones 1 al 19, donde se concentra un número cercano al 20% (29).
En una tercera parte (33%) de los pacientes con DMD/DMB, la mutación causante de la enfermedad no involucra alteraciones de tipo deleción o duplicación en la estructura del gen de la distrofina. En estos casos, el cambio de un nucleótido único o de unas pocas bases, es identificado como la causa de la mutación (30); causando el cambio de un codón original por un codón diferente que codifica para otro aminoácido ("missense"); o por el cambio de un codón que codifica para un aminoácido por un codón que codifica para una secuencia de terminación o de parada ("nonsense"), lo que resulta en una secuencia de tamaño diferente a la original o con una información genética distinta que no corresponde a la presente en la cadena original.
Correlación genotipo-fenotipo
Para establecer la correlación entre el cuadro clínico de DMD y DMB y los hallazgos moleculares se han propuesto dos hipótesis: la hipótesis del corrimiento del marco de lectura (CMLT) y la hipótesis del daño en la membrana (DM).
Cuando se trata de deleciones o inserciones que involucran unos pocos pares de bases (que no son múltiplo de tres), se altera el marco de lectura traduccional a partir del punto donde ocurre la mutación ("frameshift"); así, la secuencia resultante de aminoácidos será diferente en la región carboxiterminal de la proteína; estas mutaciones son llamadas mutaciones por fuera del marco de lectura ("out of frame") y, al parecer, están asociadas a una mayor severidad de la enfermedad.
Por otra parte, cuando el número de bases insertadas o perdidas es un múltiplo de tres, se dice que la distrofina está dentro del marco de lectura traduccional("in frame"); lo que quiere decir que aunque se remuevan grandes fragmentos de la secuencia normal del gen, el marco de lectura se conserva. Estas mutaciones "in frame" están asociadas generalmente a pacientes con DMB (23, 29, 30).
Para evaluar los fenómenos "out frame" e "in frame" de las mutaciones que causan CMLT, se clasificaron los bordes de los exones en tres tipos (1, 2 y 3), de acuerdo con la posición de los tripletes codificantes, una deleción que une dos exones con bordes del mismo tipo conserva el marco de lectura; por el contrario, una mutación que reúne dos exones con tipos de borde diferentes modifica el marco de lectura, conduciendo a la terminación temprana de la proteína o a una proteína anormal, dado que el cambio en la secuencia de aminoácidos puede generar mutaciones del tipo "missense" o "nonsense" (31). La gran mayoría de mutaciones del tipo deleción, que afectan el marco de lectura traduccional, comprometen el dominio distal rod o remueven la región aminoterminal de la proteína y se relacionan con un fenotipo severo de la enfermedad (DMD); en estos pacientes, generalmente, no se encuentra distrofina en la membrana del sarcolema (32).
Se ha establecido una correspondencia entre las deleciones que ocurren en diferentes segmentos del gen de la distrofina y su efecto sobre el marco de lectura traduccional, con los fenotipos clínicos de los pacientes sea este DMD o DMB. Deleciones dentro de la región aminoterminal (exones 1 al 8) de la proteína, resultan en un rango de fenotipos que van del leve (DMB) al severo (DMD), lo cual sugiere que la habilidad para unirse a la actina es muy importante, pero no absolutamente indispensable. Las deleciones en el dominio central rod (exones 10 al 60) que conservan el marco de lectura generalmente están asociadas al fenotipo menos severo, DMB. Las mutaciones en el dominio rico en cisteína (exones 65 al 67), o que afectan la primera mitad del dominio carboxiterminal (exones 68 al 70), siempre resultan en el fenotipo severo, DMD, hallazgo que indica la importancia de estas regiones del gen para el normal funcionamiento de la distrofina (33).
En cuanto a la hipótesis del daño a la membrana (DM), éste propone la pérdida de la función de la proteína distrofina, la cual, normalmente esta unida a la membrana muscular y ayuda así a mantener la integridad de la fibra muscular. En la ausencia de la distrofina, las fibras degeneran y se vuelven susceptibles de morir con la contracción muscular. De este modo, de acuerdo con la severidad del daño o la disfunción de la proteína, se verán diferentes grados de funcionalidad que estará directamente relacionada con la severidad de la enfermedad.
Métodos de análisis molecularen DMD/DMB
Dentro de los métodos de biología molecular, se pueden mencionar varias técnicas disponibles para el análisis del ADN, del ARN o de las proteínas, cada una de ellas con ventajas y desventajas en razón de costo, sencillez y eficiencia; algunas de estas técnicas son: reacción en cadena de la polimerasa (PCR) múltiple, RT-PCR y polimorfismos conformacionales de cadena sencilla, entre otras.
De los diferentes métodos de análisis molecular de mutaciones del gen de la distrofina arriba enunciados, el método de PCR es de amplia aceptación porque permite caracterizar de manera rápida y precisa el 98% de las mutaciones de tipo deleción o duplicación del gen, al analizar 18 regiones del gen que son coamplificadas en tres o cuatro reacciones individuales de PCR (34, 35). Los productos amplificados por PCR multiplex son corridos en geles de agarosa al 2% y teñidos con bromuro de etidio (36, 37), para la visualización de los fragmentos resultantes de la amplificación de cada uno de los exones. La ausencia de uno a más de éstos, en comparación con un control sano, permite identificar con mucha facilidad una deleción (23, 36, 37) (Figura 4).
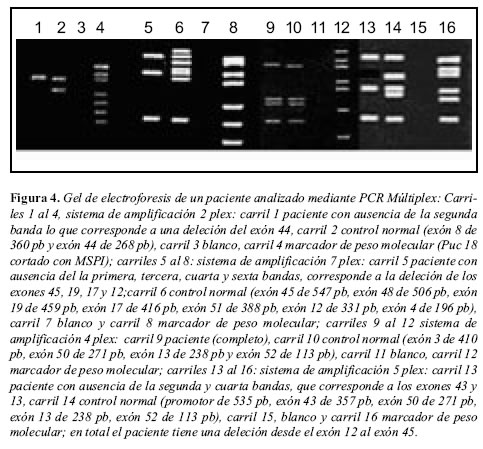
Genética y asesoramiento genético
Por ser una enfermedad de herencia ligada al sexo recesiva, en los casos donde hay dos o más afectados, también llamados casos familiares, al menos la mitad de las mujeres hermanas de un afectado y todas sus hijas serán portadoras obligatorias de la enfermedad, la cual segregarán nuevamente a la mitad de sus hijos varones (afectados) y a la mitad de sus hijas (portadoras) (34). Estas familias pueden ser objeto de acciones de prevención primaria y secundaria mediante el asesoramiento genético y el diagnóstico prenatal o algunas acciones reproductivas como la donación de oocitos, el diagnóstico preimplantación o la adopción que pueden evitar el nacimiento de nuevos niños afectados.
Conclusión
En conclusión, las DMD y DMB, la distrofia muscular más frecuente en el hombre, son formas alélicas debidas a la mutación de un mismo gen, llamado distrofina y localizado en el brazo corto del cromosoma X. El gen de la distrofina establece, mediante la asociación a varias proteínas, un enlace que estabiliza el citoesqueleto con el exterior de la membrana del sarcolema y es una proteína que se expresa en diferentes tejidos y órganos. Las mutaciones del gen de la distrofina son heterogéneas, sin embargo, la mayor parte de los afectados exhiben la ausencia de uno o más exones del gen en dos regiones proclives a alterarse. La extensión y localización de cada mutación genera alteraciones en la capacidad de la distrofina de asociarse con otras proteínas, lo que ocasiona disfunción de las miofibrillas causando tanto la forma severa (DMD) como leve (DMB) de esta distrofia muscular. Si bien existen métodos variados para el análisis del gen, a nivel de ADN o ARN y/o la proteína, el método de elección es el análisis mediante reacción en cadena de la polimerasa (PCR). El asesoramiento genético, el diagnóstico prenatal y el ofrecimiento de opciones reproductivas pueden ser herramientas útiles para la prevención de nuevos afectados en las familias.
Referencias
1. Emery AE. The muscular dystrophies. BMJ 1998; 317:991-5. [ Links ]
2. Verellen-Dumoulin C, Freund M, De Meyer R, Laterre C, Frederic J, Thompson MW, et al. Expression of an X-linked muscular dystrophy in a female due to translocation involving Xp21 and non-random inactivation of the normal X chromosome. Hum Genetics 1984; 67:115-9. [ Links ]
3. Francke U, Ochs HD, de Martinville B, Giacalone J, Lindgren V, Disteche C, et al. Minor Xp21 chromosome deletion in a male associated with expression of Duchenne muscular dystrophy, chronic granulomatous disease, retinitis pigmentosumn and McLeod syndrome. Am J Hum Genet 1985; 37:250-67. [ Links ]
4. Anderson MS, Kunkel LM. The molecular and biochemical basis of Duchenne muscular dystrophy. Trends Biochem Sci 1992; 17:289-92. [ Links ]
5. Tyler KL. Origins And Early Descriptions Of Duchenne Muscular Dystrophy. Muscle Nerve 2003; 28:40222. [ Links ]
6. Roberts RG. Dystrophins and dystrobrevins. Genome Biol 2001; 2:17. [ Links ]
7. Duchenne GB. Recherches sur la paralysie musculaire pseudo-hypertrophique ou paralysie myo-sclérosique. Archives Generáles de Médicine 1868; 11:552-88. [ Links ]
8. Gowers WR. Pseudo-hypertrophic muscular paralysis. Londres, Ed Churchill. 1879. [ Links ]
9. Escobar-Cedillo RE, Miranda A, Lona S, Castillo M. Análisis del patrón de interferencia en pacientes con distrofia muscular. Rev Neurol 2004; 39:517-20. [ Links ]
10. Buchthal F. An Introduction to electromyography. Copenhagen: Scandinavian University Books; 1957. [ Links ]
11. Dorfman LJ, McGill KC. AAEE Minimonograph # 29: Automatic quantitative electromyography. Muscle Nerve 1988; 11:804-18. [ Links ]
12. Cullen MJ. Morphological changes in dystrophic muscle. Br Med Bull 1980; 36:145-52. [ Links ]
13. Niebroj-Dobosz I, Fidzianska A, Hausmanowa-Petrosewiez I. Controversies about the functional dystrophin in muscle. Folia Neuropathol 2001; 39:253-8. [ Links ]
14. Zubrzycka-Gaarn EE, Bulman DE, Karpati G, Burghes AH, Belfall B, Klamut HJ, et al. The Duchenne muscular dystrophy gene product is localized in the sarcolemma of human skeletal muscle. Nature 1988; 333:466-69. [ Links ]
15. Sciandra F, Bozzi M, Bianchi M, Pavoni E, Giardina B, Brancaccio A. Dystroglycan and muscular dystrophies related to the dystrophin-glycoprotein Complex. Ann Ist Super Sanita 2003; 39:173-81. [ Links ]
16. Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletion of the DMD lucus. Genomics 1988; 2:90-5. [ Links ]
17. Malhotra SB, Hart KA, Klamut HJ, Thomas NS, Bodrug SE, Burghes AH, et al. Frame-shift deletions in patients with Duchenne and Becker muscular dystrophy. Science 1988; 242:755-9. [ Links ]
18. Worton R, Gillard E. Duchenne Muscular Dystrophy en Molecular Basis of Neurology. Boston Blackwell Scientific Publications. 1992. Capítulo 3. [ Links ]
19. Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoeskeletal protein. Cell 1988; 53:219-26. [ Links ]
20. Nishio H, Takeshima Y, Narita N, Yanagawa H, Suzuki Y, Ishikawa Y, et al. Identification of a novel first exon in the human dystrophin gene and of a new promoter located more than 500 Kb Upstream of the nearest known promoter. J Clin Invest 1994; 94:1037-42. [ Links ]
21. Prieto JC, Keyeux G, Camacho M. Caracterización de mutaciones en pacientes con distrofia muscular de Duchenne y Becker. Tesis Pontificia Universidad Javeriana 1997: 24-39. [ Links ]
22. Den Dunnen JT, Grootscholten PM, Bakker E, Blonden LA, Ginjaar HB, Wapenaar MC, et al. Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am J Hum Genet 1989; 45:835-47. [ Links ]
23. Silva CT, Fonseca D, Restrepo CM, Contreras NC, Mateus HD. Deleciones en el gen de la distrofina en 62 familias colombianas: correlación genotipo fenotipo para la distrofia muscular de Duchenne y Becker. Colomb Med 2004; 35:191-8. [ Links ]
24. Silva E. Informe de casos de distrofia muscular de Duchenne y Becker. En Informe Epidemiológico Nacional 1998; 3:69-72. [ Links ]
25. Baranzini SE, Giliberto F, Dalamon V, Barreiro C, Garcia-Erro M, Grippo J, et al. Carrier detection in Duchenne and Becker muscular dystrophy Argentine families. Clin Genet 1998; 54:503-11. [ Links ]
26. Delgado LW, Pineda-Del VL, Borjas L, et al. Molecular diagnosis of Duchenne/Becker muscular dystrophy in Venezuela patients with the polymerase chain reaction. Clin Invest 1994; 35:195-207. [ Links ]
27. Keyeux G, Rodas C, Bienvenu T, Garavito P, Vidaud D, Sanchez D, et al. CFTR Mutations in patients from Colombia: Implications for local and regional Molecular Diagnosis Programs. Hum Mutat 2003; 22:59. [ Links ]
28. Hoffman EP, Kunkel LM. Dystrophin abnormalities in Duchenne/Becker muscular Dystrophy. Neuron 1989; 2:1019-29. [ Links ]
29. Shomrat R, Gluck E, Legum C, Shiloh Y. Relatively low proportion of dystrophin gene deletions in Israeli Duchenne and Becker muscular dystrophy patients. Am J Med Genet 1994; 49:369-73. [ Links ]
30. Yau SC, Roberts RG, Borrow M, Mathew CG. Direct diagnosis of carriers of point mutations in Duchenne muscular dystrophy. The Lancet 1993; 341:273-75. [ Links ]
31. Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet 1989; 45:498-506. [ Links ]
32. Beggs AH, Hoffman EP, Snyder JR, Arahata K, Specht L, Shapiro F, et al. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am J Hum Genet 1991; 49:54-67. [ Links ]
33. Tinsley JM, Blake DJ, Zuellig RA, Davies KE. Increasing complexity of the dystrophin-associated protein complex. Proc Natl Acad Sci USA 1994; 91:8307-13. [ Links ]
34. Kitoh Y, Matsuo M, Nishio H, Takumi T, Nakajima T, Masumura T, et al. Amplification of ten deletion-rich exons of the dystrophin gene by polymerase chain reaction shows deletions in 36 of 90 Japanese families with Duchenne muscular dystrophy. Am J Med Genet 1992; 42:453-7. [ Links ]
35. Chamberlain JS, Gibbs RA., Rainer JE, Caskey CT. Multiplex PCR for the diagnosis of Duchenne Muscular Dystrophy. En: Innis M, Gelfond D, Sninsky J, White J, eds, PCR protocols: A guide to Methods and Applications. Cambridge: Academic Press; 1990.p.273-281. [ Links ]
36. Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acid Res1988; 16:1141-56. [ Links ]
37. Bassam BJ, Caetano-Anolles G, Gresshoff PM. Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal Biochem 1991; 196: 80-3. [ Links ]
38. Hernández P, Gomez YM, Silva CT, Restrepo CM. Identificación de portadoras de Distrofia Muscular de Duchenne y Becker (DMD/DMB) mediante análisis de dosis génica y polimorfismos de DNA. Biomédica 2000; 20:228-237. [ Links ]

