










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.30 no.3 Bogotá July/Sept. 2005
Dr. Carlos Olimpo Mendivil A.: Profesor Adjunto e Investigador, División de Lípidos y Diabetes, Facultad de Medicina, Universidad Nacional de Colombia, Bogotá, D.C.
Recibido: 15/08/05 Aprobado: 01/09/05
Resumen
El síndrome metabólico es un conjunto de alteraciones que se asocian a la resistencia a la insulina. El tejido adiposo, como órgano endocrino activo, produce varios mediadores que afectan directa e indirectamente la sensibilidad a la insulina en varios tejidos. En la presente revisión se examinan los mecanismos fisiológicos de la acción insulínica, y la forma como se pueden ver afectados por la acción de varias hormonas de origen adipocitario, incluyendo leptina, factor de necrosis tumoral alfa y adiponectina.
Palabras clave: síndrome metabólico, adipocito, leptina, adiponectina, hipertensión.
Summary
Metabolic syndrome comprises a series of alterations associated with insulin resistance. The adipose tissue, as an active endocrine organ, produces several mediators that act both directly and indirectly on insulin sensitivity in several tissues. This paper is designed to review the physiological mechanisms of insulin activity and the way in which they may be affected by the action of several hormones originating in the adipose tissue, including leptin, tumor necrosis factor alpha and adiponectin.
Key words: metabolic syndrome, adipocyte, leptin, adiponectin, hypertension.
Introducción
El síndrome metabólico es un conjunto de alteraciones presentes en diferentes sistemas orgánicos, pero asociadas todas a un mismo fenómeno fisiopatológico: la resistencia a la insulina.
Se define resistencia a la insulina como una respuesta tisular inferior a la esperada, en presencia de concentraciones normales o supranormales de insulina. Por supuesto, la resistencia a la insulina puede darse en todos los tejidos blanco de la insulina; sin embargo, los actores más importantes en la génesis del síndrome metabólico son: tejido adiposo, músculo esquelético, hígado, páncreas y cerebro.
Para aproximarnos a la compleja interrelación entre estos órganos y así comprender la relación entre obesidad y síndrome metabólico, tocaremos los siguientes puntos:
1. Adiposidad y componentes clave del síndrome metabólico:
- Adiposidad y tensión arterial
- Adiposidad y lípidos séricos
- Adiposidad y metabolismo de carbohidratos
2. ¿Cómo actúa la insulina?
3. ¿Cómo puede la adiposidad afectar la acción de la insulina?
Adiposidad y componentes clave del síndrome metabólico
Adiposidad y tensión arterial
A nivel poblacional, existe una relación directa y lineal entre el índice de masa corporal (IMC) y el riesgo de desarrollar hipertensión arterial, tanto en hombres como en mujeres (1). Algunos de los posibles nexos que explican la asociación entre peso corporal y tensión arterial están dados a través de la resistencia a la insulina: el incremento en la adiposidad se acompaña de mayor secreción de hormonas que promueven la resistencia a la insulina (que se revisarán más adelante), y la resistencia a la insulina ocasiona elevación de la tensión arterial por:
· Menor efecto vasodilatador de la insulina (2).
· Retención de agua y sodio, como consecuencia de la hiperinsulinemia. Al parecer a pesar de la resistencia a la insulina en otros tejidos, los efectos renales de la hormona están preservados (3).
· La resistencia a la insulina se acompaña de disfunción endotelial, lo que ocasiona una menor biodisponibilidad de óxido nítrico y por tanto un tono vasoconstrictor (4).
· Las neuronas del tallo cerebral que regulan el tono simpático eferente hacia las arteriolas (principal determinante de la resistencia vascular periférica), son inhibidas por neuronas cuyos somas están en el hipotálamo y que a diferencia de las otras neuronas, poseen unan captación adaptativa de glucosa, estimulada por la insulina. Cuando hay resistencia a la insulina e hiperinsulinemia, las neuronas hipotalámicas captan más glucosa, y eso hace que disminuya la actividad inhibitoria hacia las neuronas del tallo cerebral. El efecto final es que se incrementa la actividad simpática por desinhibición de las neuronas simpáticas del tallo cerebral (5).
Adiposidad y lípidos séricos
Es bien conocido que el sobrepeso se asocia a niveles elevados de triglicéridos (TG) y niveles disminuidos de colesterol de HDL en sangre (6). Al parecer el nexo se encuentra en la resistencia a la insulina:
· El individuo con cierta predisposición genética y expuesto a un estilo de vida que lo favorece, gana peso y adiposidad.
· Los adipocitos se hacen más grandes y resistentes a la insulina, liberando además mediadores locales que inducen resistencia a la insulina en otras células.
· Como consecuencia de la falta de acción insulínica, la lipoproteinlipasa (LPL-1), presente en el endotelio y encargada de hidrolizar los TG de las VLDL y los quilomicrones, pierde actividad.
· Los TG del plasma no son degradados ni almacenados en tejido adiposo sino que siguen circulando de ahí la hipertrigliceridemia.
· La hipertrigliceridemia activa excesivamente a la PTEC (proteína de transferencia de ésteres de colesterol), que transfiere colesterol de las HDL - LDL a las VLDL quilomicrones, y transfiere TG de las VLDL quilomicrones a las LDL - HDL.
· El sobrepeso ha conducido a que las lipoproteínas ricas en TG ahora sean ricas en colesterol, y las lipoproteínas ricas en colesterol, ahora sean ricas en TG.
· Debido a la acción de la lipasa hepática, las LDL y HDL pierden los TG que habían recibido.
· Las HDL son ahora más pobres en colesterol; de ahí el cHDL bajo.
· Las LDL son más pequeñas y proporcionalmente más ricas en proteína; de ahí las ldl pequeñas y densas.
Adiposidad y metabolismo de carbohidratos
El incremento en la adiposidad corporal afecta negativamente la acción insulínica debido a la producción de hormonas inductoras de resistencia a la insulina, y deteriora la función de las células beta pancreáticas por inducción de elevación de ácidos grasos libres y lipotoxicidad, así como hiperleptinemia. Los mecanismos de los efectos deletéreos de la adiposidad excesiva sobre las células beta y sobre la acción de insulina se desarrollan más adelante.
¿Cómo actúa la insulina?
Una vez secretada por las células beta del páncreas, la insulina se distribuye a sus tejidos blanco y se une a su receptor en la membrana celular. El receptor de insulina es un receptor con actividad tirosín-cinasa intrínseca, que consta de cuatro subunidades: dos subunidades alfa transmembranales y dos subunidades beta intracitoplásmicas (7). La estructura del receptor de insulina se resume en la Figura 1.
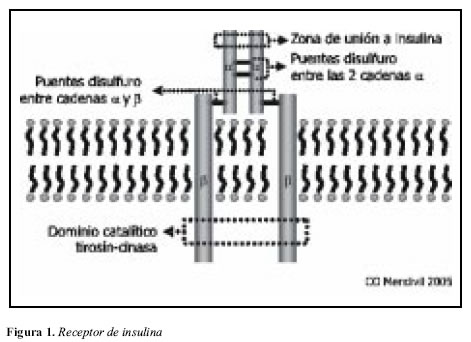
El receptor de insulina puede considerarse una enzima alostérica, con dos subunidades regulatorias (las subunidades alfa), que mantienen inhibidas a dos subunidades catalíticas (las subunidades beta). Cuando la insulina se une a las subunidades alfa, el efecto inhibitorio de éstas cesa y las subunidades beta ejercen su acción catalítica, transfosforilándose entre sí, y fosforilando en tirosina a un grupo de proteínas conocidas como los sustratos del receptor de insulina (IRS, por Insulin Receptor Substrates).
Se conocen cuatro proteínas IRS, de las cuales las más extensamente estudiadas son IRS-1 e IRS-2. IRS-1 es una proteína rica en regiones de unión a tirosinas fosforiladas (regiones PTB, de PhosphoTyrosine Binding), que le permiten unirse al receptor y ser fosforilado por él en sus residuos de tirosina (8). Una vez el IRS-1 es fosforilado, liga y recluta a la membrana celular a dos moléculas de gran importancia en la respuesta biológica a la insulina: PI3K y Grb-2; lo que inicia una cascada de señalización intracelular que culmina con la expresión de todos los efectos fisiológicos de la insulina (9).
Los efectos de la insulina se pueden clasificar en dos grandes grupos: efectos a corto plazo o "metabólicos" y efectos a mediano y largo plazos o "tróficos". Los principales efectos del primer grupo son:
· Estímulo a la captación de glucosa, mediante el favorecimiento de la traslocación de los glucotransportadores GLUT-4 a la membrana plasmática en músculo y tejido adiposo (10).
· Estímulo a la síntesis de glucógeno e inhibición de su degradación en hígado y músculo (10).
· Estímulo al metabolismo oxidativo de la glucosa (glucólisis) (10).
· Inhibición de la gluconeogénesis hepática (10).
· Estímulo a la captación y almacenamiento de grasas por el tejido adiposo (estímulo a la LPL-1 y triglicérido sintasa) (10).
· Inhibición de la lipólisis en tejido adiposo (por inhibición de la lipasa adipolítica u hormonosensible) (10).
Los principales efectos a intermedio y largo plazos o "tróficos" de la insulina son:
· Efectos sobre la captación/retención de iones y el metabolismo hidroelectrolítico (11).
· Estímulo a la síntesis e inhibición de la degradación de proteínas (11).
· Efectos sobre la expresión génica (transcripción) (11).
· Efectos sobre el recambio del mRNA (11).
· Estímulo al crecimiento, proliferación y diferenciación celulares (11).
¿Cómo puede la adiposidad afectar la acción de la insulina?
El tejido adiposo, más allá de ser un depósito inerte de TG, es un tejido endocrino sumamente activo de cuyo correcto funcionamiento depende el equilibrio metabólico de todo el organismo. Cuando un individuo tiene un balance energético positivo sostenido en el tiempo, ese excedente calórico se almacena inicialmente como TG en el tejido adiposo. Los adipocitos, más que dividirse y generar nuevas células, almacenan más TG por célula, lo que genera adipocitos más grandes (12). La hipertrofia del tejido adiposo hace que los adipocitos tengan características funcionales particulares:
· Menor concentración de receptores de insulina por unidad de área de membrana plasmática, por tanto son menos sensibles a la insulina (13).
· Recordemos que en el interior de los adipocitos se encuentra la enzima lipasa adipolítica, encargada de hidrolizar los TG adipocitarios a glicerol y ácidos grasos libres e inhibida por acción de la insulina. Los adipocitos grandes, al ser menos sensibles a la insulina, tienen una mayor actividad de lipasa adipolítica, y por tanto producen constantemente ácidos grasos libres, que son vertidos a la circulación (12).
· Tienen un patrón especial de secreción endocrina: producen una mayor cantidad de leptina y factor de necrosis tumoral alfa (TNF-alfa), y una menor cantidad de adiponectina (14,15).
Leptina
La leptina es una hormona peptídica que es estructuralmente similar a las citocinas, y se produce en proporción a la masa de tejido adiposo. La producción de leptina es mayor en el tejido adiposo subcutáneo que en el visceral (16).
Existen receptores para leptina en el hipotálamo, encargados de regular el apetito y el gasto energético, pero también se expresan receptores de leptina en músculo y célula beta del páncreas (16). En las células beta del páncreas, la unión de la leptina a su receptor ocasiona un aumento en la producción de un grupo de proteínas llamadas SOCS (Supressors Of Cytokine Signaling), especialmente SOCS3 (17). La proteína SOCS3 se une a uno de los sustratos del receptor de insulina (IRS-2) y ocasiona su ubiquitinación (la ubiquitina funciona como un rótulo para las proteínas que deben ser degradadas), IRS-2 es degradado y la célula beta se queda sin uno de sus más importantes factores de supervivencia, lo que conduce a su apoptosis. Ese es uno de los mecanismos por los cuales el incremento en la adiposidad propicia la disminución en la masa de células beta y favorece el desarrollo de prediabetes y diabetes (Figura 2).
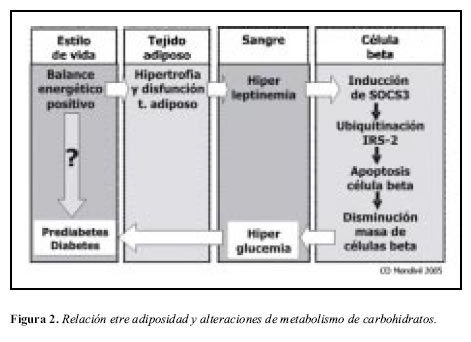
La leptina es una señal de adiposidad, que funciona como un estímulo al "lipostato" hipotalámico indicando que ya hay suficiente tejido adiposo y es hora de reducir la ingesta e incrementar el gasto calórico.
Cuando se descubrieron la leptina y su receptor en 1994-96 se pensó que la suplementación externa de leptina podría ser una alternativa terapéutica en el manejo de la obesidad. Sin embargo, cuando se midieron los niveles de leptina en humanos obesos, los niveles estaban usualmente incrementados (18), indicando que el problema no era deficiencia de leptina.
Así pues, el principal efecto de la leptina sobre la acción de insulina es reducirla, pero no generando resistencia a la insulina sino afectando su producción pancreática.
TNF-alfa
El TNF-alfa, inicialmente identificado en macrófagos y llamado caquectina, es producido también por el tejido adiposo y cumple esencialmente una función paracrina (sobre células adyacentes) y autocrina (sobre el propio tejido adiposo) (19). Existen dos tipos de receptores de TNF-alfa, y el tejido adiposo expresa ambos (20).
En algunos estudios se ha encontrado correlación entre los niveles plasmáticos de TNF-alfa y la resistencia a la insulina, pero en otros no. Sin embargo, ésta aparente contradicción puede deberse a que sólo una pequeña fracción del TNF-alfa que es secretado sale a la circulación general, la mayor parte cumple su acción en el propio tejido adiposo y es degradado in situ (20).
El TNF-alfa inhibe las enzimas involucradas en la captación de ácidos grasos, en la captación de glucosa y en la síntesis de triglicéridos, causando por tanto hiperglucemia e incremento en la concentración de ácidos grasos libres en sangre. Cuando TNF-alfa se une a su receptor en hígado, se estimula la síntesis de colesterol y ácidos grasos, y tanto en tejido adiposo como en hígado, causa resistencia a la insulina, pues activa serina-cinasas (isoenzimas atípicas de proteincinasa C - PKC) que compiten con el receptor de insulina por la fosforilación de sus sustratos (IRS-1 y 2) (10).
Cuando los IRS son fosforilados en serinas y treoninas en lugar de tirosinas, toda la cascada de señalización posterior se afecta y la respuesta efectora a la insulina se inhibe.
El TNF-alfa es, por tanto, la auténtica hormona de la resistencia a la insulina.
Adiponectina
La adiponectina es una hormona similar a las proteínas del complemento, que es producida específicamente por el tejido adiposo y que, a diferencia del TNF-alfa, llega a la circulación sistémica en su mayor parte (21-23). Al igual que leptina, se expresa más en tejido adiposo subcutáneo que en tejido adiposo visceral, y su concentración se incrementa cuando la sensibilidad a la insulina mejora.
Para que tenga adecuada actividad biológica, la adiponectina debe estar hidroxilada y glucosilada, lo cual genera varias isoformas de acuerdo con el grado de hidroxilación y glucosilación. Adicionalmente, los monómeros de adiponectina forman trímeros mediante enlaces disulfuro, dos de estos trímeros se agregan para formar un hexámero, y muchos hexámeros se unen para formar adiponectina de alto peso molecular (24).
Se han identificado dos receptores diferentes de adiponectina: el receptor AdipoR1, que se expresa primordialmente en músculo, y el AdipoR2, que se expresa primordialmente en hígado (25). El Adipo R2 tiene mayor afinidad por la adiponectina completa (full-lenght), mientras que el AdipoR1 tiene mayor afinidad por una forma corta de adiponectina generada por proteólisis.
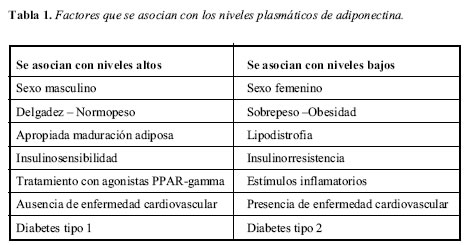
Los niveles plasmáticos de adiponectina guardan una fuerte relación inversa con el peso corporal, y una fuerte relación directa con la sensibilidad a la insulina, como se ha demostrado en estudios epidemiológicos y en estudios con intervención farmacológica para mejorar la sensibilidad a la insulina (26). Diversos factores influencian los niveles plasmáticos de adiponectina, tal como se resume en la Tabla 1 (24).
La adiponectina se caracteriza por poseer efectos biológicos que se podrían llamar "protectores" o "antiaterogénicos":
· Reduce la producción hepática de glucosa (23, 24).
· Estimula la betaoxidación de ácidos grasos en hígado (23, 24).
· Inhibe la adhesión de monocitos al endotelio vascular (23, 24).
· Inhibe la expresión de receptores basurero ("scavenger") de LDL en los macrófagos (23, 24).
· Inhibe la proliferación y migración de células musculares lisas en la pared arterial (23, 24).
· Incrementa la fosforilación del receptor de insulina, y por ende todos los demás efectos insulínicos (23, 24).
En ratones transgénicos ob/ob (sin capacidad de producir leptina), la sobreexpresión de adiponectina induce reducción de triglicéridos plasmáticos, normalización de la glucemia e insulinemia pero, algo muy interesante, el peso corporal se incrementa aún más y llegan a tener niveles de adiposidad sorprendentes (24). Es por este perfil de efectos que se ha comparado al efecto de la adiponectina con el efecto del tratamiento con agonistas PPAR-gamma.
Como hemos podido observar, la obesidad media sus efectos deletéreos como potenciadora del síndrome metabólico primordialmente a través de la inducción de resistencia a la insulina. La lucha contra la obesidad mediante medidas de salud pública, y el desarrollo de nuevos agentes que contrarresten las hormonas adipocitarias nocivas, abre nuevas expectativas en la prevención de la diabetes y las enfermedades cardiovasculares.
Referencias
1. Bray GA. Medical consequences of obesity. J Clin Endocrinol Metab 2004; 89:2583-89. [ Links ]
2. Anderson EA, Mark AL. The vasodilator action of insulin: implications for the insulin hypothesis of hypertension. Hypertension 1993; 21:136-41. [ Links ]
3. Reaven GM. The kidney: an unwilling accomplice in syndrome X. Am J Kidney Dis 1997; 30:928-31. [ Links ]
4. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005; 365:1415-28. [ Links ]
5. Reaven GM, Lithell H, Landsberg L. Hypertension and associated metabolic abnormalities - the role of insulin resistance and the sympathoadrenal system. N Eng J Med 1996; 334:374-81. [ Links ]
6. Howard BV, Ruotolo G, Robbins DC. Obesity and dyslipidemia. Endocrinol Metab Clin North Am 2003; 32:855-67. [ Links ]
7. Ullrich A, Bell JR, Chen EY, Herrera R, Petruzzelli LM, Dull TJ, et al. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 1985; 313:756-61. [ Links ]
8. Giovannone B, Scaldaferri ML, Federici M, Porzio O, Lauro D, Fusco A, et al. Insulin receptor substrate (IRS) transduction system: distinct and overlapping signaling potential. Diabetes Metab Res Rev 2000; 16:434-41. [ Links ]
9. Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-Kinase regulates cell signaling and survival. Mol Cell Biol 2002; 22:965-77. [ Links ]
10. Pirola L, Johnston AM, Van Obberghen E. Modulation of insulin action. Diabetologia 2004; 47:170-84. [ Links ]
11. Flakoll PJ, Carlson MG, Cherrington AD. Acción fisiológica de la insulina. En: Le Roith D, Taylor SI, Olefsky JM, eds. Diabetes Mellitus, Fundamentos y Clínica. 2 edición. México, Mc Graw Hill, 2003. PAGINAS? [ Links ]?
12. Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO rep 2001; 2:282-6. [ Links ]
13. Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes Relat Metab Disord 2003; 27:S535 [ Links ]
14. Hotamisligil GS. Molecular mechanisms of insulin resistance and the role of the adipocyte. Int J Obes Relat Metab Disord 2000; 24:S23-7. [ Links ]
15. Fusshauer M, Paschke R. Regulation of adipocytokines and insulin resistance. Diabetologia 2003; 46:1594-03. [ Links ]
16. Van Harmelen V, Reynisdottir S, Eriksson P, Thorne A, Hoffstedt J, Lonnqvist F, et al. Leptin secretion from subcutaneous and visceral adipose tissue in women. Diabetes 1998; 47:91317. [ Links ]
17. Brady MJ. IRS2 takes center stage in the development of type 2 diabetes. J Clin Invest 2004; 114:886-8. [ Links ]
18. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature 1998; 95:76370. [ Links ]
19. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993; 259:8791. [ Links ]
20. Ruan H, Lodish HF. Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor. Cytokine Growth Factor Rev 2003; 14:44755. [ Links ]
21. Tsao TS, Lodish HF, Fruebis J. ACRP30, a new hormone controlling fat and glucose metabolism. Eur J Pharmacol 2002; 440:21321. [ Links ]
22. Matsuzawa Y, Funahashi T, Kihara S, Shimomura I. Adiponectin and metabolic syndrome. Arterioscler Thromb Vasc Biol 2004; 24:2933. [ Links ]
23. Chandran M, Phillips SA, Ciaraldi T, Henry RR, et al. Adiponectin: more than just another fat cell hormone?. Diabetes Care 2003; 26:244250. [ Links ]
24. Scheffer P. American Diabetes Association Scientific Sessions Outstanding Scientific Achievement Lecture 2005: Adipose tissue, From fat storage to endocrine organ. [ Links ]
25. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetics metabolism effects. Nature 2003; 423:7629. [ Links ]
26. Diez JJ, Iglesias P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur J Endocrinol 2003; 148:293300. [ Links ]

