










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.31 no.1 Bogotá Jan./Mar. 2006
Reporte de caso y revisión
Case report and review
Dres. Juan Esteban Gómez, Luis Fernando Pava, Pablo Perafán, Marisol Badiel, María Fernanda Villegas, Camilo Arana, Jorge Guillermo Velásquez, Juan José Arango: Servicios de Electrofisiología, Hemodinamia Cardiaca en el Instituto de Investigaciones Clínicas, Fundación Valle del Lili. Cali, Colombia.
Correspondencia: Dr. Luis Fernando Pava Molano, Fundación Valle del Lili, Unidad de Electrofisiología, Carrera 98 No. 18-49, Cali - Colombia, Tel. 2-3319090, Fax. 2-3157974. E-mail: fpava@hotmail.com
Recibido: 13/12/05 Aceptado: 15/03/06
Resumen
La prolongación anormal del intervalo QT tiene relevancia por su asociación con inestabilidad eléctrica ventricular, pudiéndose presentar arritmias ventriculares polimórficas en "puntas torcidas". Los síntomas acompañantes de estas arritmias son el síncope, convulsiones o la muerte súbita. Se puede clasificar en síndrome de QT prolongado congénito, el cual se hereda o lo adquiere el individuo de manera esporádica y el síndrome adquirido que se puede presentar asociado a fármacos u otras situaciones clínicas. Presentamos el caso de una mujer de 41 años de edad que desarrolló cambios típicos de síndrome de QT prolongado por isquemia miocárdica y hacemos una breve revisión.
Palabras clave: isquemia miocárdica, QT prolongado.
Abstract
The abnormal QT interval prolongation has relevance for its association with ventricular electric instability, which could present polymorphic ventricular arrhythmias in "torsade de pointes". The underlying symptoms of these arrhythmias are syncope, convulsions or sudden death. It can be classified as congenital long QT syndrome which is inherited or the subject acquired it in a sporadic way; and the acquired syndrome that can be associated to drugs or other clinic factors. We show a case of a 41 years old woman who developed typical changes of long QT syndrome by myocardial ischemia and we make a brief review of it.
Key words: myocardical ischemia, prolonged QT.
Presentación del caso
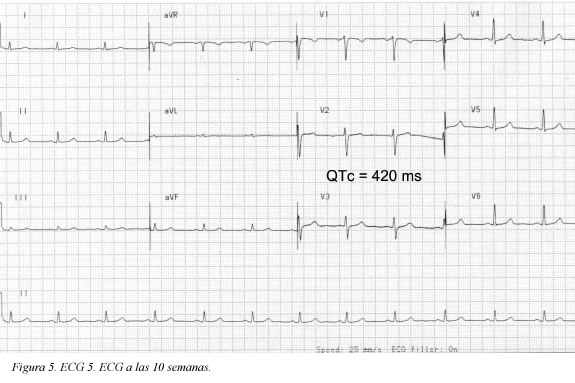
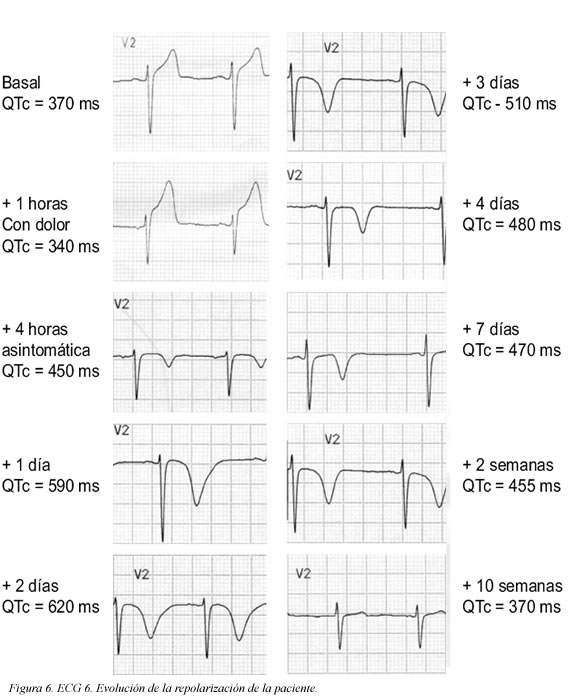
Se trata de una paciente de 41 años de edad, fumadora de veinte paquetes/año cigarrillos, sin antecedentes patológicos, personales ni familiares, quien consultó al servicio de urgencias por haber presentado un dolor torácico coronario típico de cinco minutos de duración; en el momento de la consulta la paciente estaba asintomática (ECG Figura 1); durante la primera hora de observación presentó nuevo episodio de dolor torácico que duró aproximadamente cinco minutos (ECG Figura 2) y cedió con nitratos; se practicó cateterismo que mostró leve ectasia de la arteria coronaria descendente anterior e imagen compatible con trombo reciente, pero con flujo TIMI III. Desde entonces, durante 10 semanas de seguimiento la paciente ha estado asintomática. El seguimiento del electrocardiograma (ECG) se muestra en las Figuras 3, 4 y 5. Antes de su ingreso la paciente no tomaba ningún medicamento y en el momento de su ingreso por urgencias, los electrolitos eran: sodio: 138 mEq/l, potasio:3.7 mEq/l (vn 3,6-5), magnesio: 1,8 (vn= 1,6-2,3) y cloro:108 mEq/l. Durante su hospitalización recibió tratamiento antiagregante, anticoagulante, enalapril, betabloqueador y estatinas y no recibió ningún medicamento reportado que prolongara el intervalo QTc. Presentamos los ECGs seriados (ECG Figuras 1, 2, 3, 4, 5, 6) en los que se observa la evolución de la repolarización cardiaca destacando la prolongación anormal del QT (ECG 6).
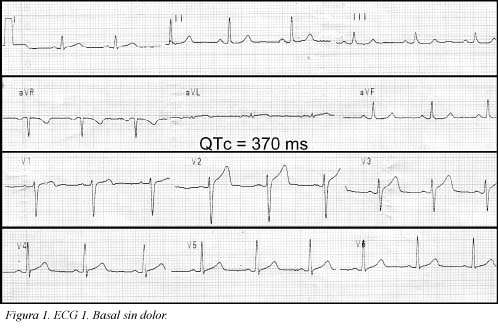
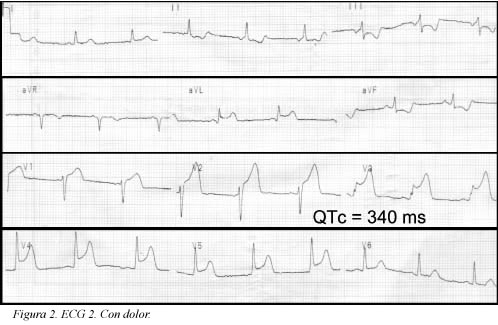
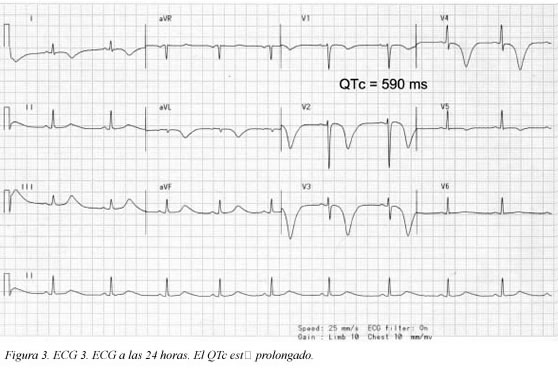
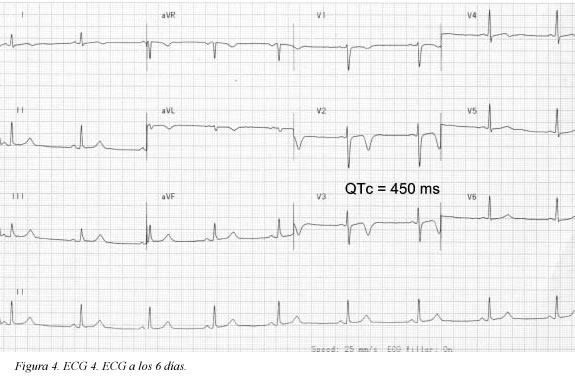
Definición
El síndrome de QT largo (SQTL) se caracteriza por la prolongación anormal de la repolarización ventricular, predisponiendo al paciente a presentar arritmias ventriculares polimórficas en "torcidas de punta", como consecuencia de oscilaciones en el potencial de membrana durante la repolarización ventricular (pospotenciales precoces), oscilaciones que reflejan inestabilidad eléctrica y que en caso de propagarse se pueden convertir en taquicardia ventricular polimórfica. Los síntomas son debidos a las taquicardias, pudiendo presentarse presíncope, síncope, palpitaciones, convulsiones o muerte súbita (1). La prolongación del intervalo QT ocasionada exclusivamente por el aumento de la duración del complejo QRS no constituye SQTL y no se asocia con mayor riesgo de arritmias ventriculares polimórficas (2). El SQTL puede ser congénito o adquirido, sin embargo, no está completamente claro que las formas secundarias sean sólo como consecuencia de un factor desencadenante (3). Se han descubierto mutaciones en genes únicos que producen diferentes síndromes, por ejemplo mutaciones en los canales del sodio causando SQTL y a la vez síndrome de Brugada; este polimorfismo añade incertidumbre sobre la verdadera existencia de una única enfermedad o sobre la posibilidad de que existan genes que interactúen con el ambiente o con otros genes reguladores, que permitan expresar la enfermedad de manera espontánea o ante la presencia de un factor desencadenante, como por ejemplo en el caso de nuestra paciente, la isquemia miocárdica (4).
SQTL congénito
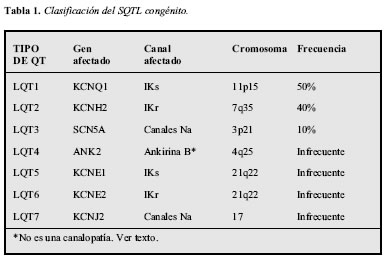
La base genética de este síndrome fue confirmada con el descubrimiento de mutaciones en los genes codificadores de la corriente rectificadora tardía cardiaca del potasio y de corrientes del sodio, KCNH2 (HERG) y SCN5A, respectivamente. En la Tabla 1 se muestran las características principales de las formas congénitas. La mayoría de casos de SQTL están relacionados con la disfunción en los canales de potasio o con las fallas de los canales del sodio para inactivarse de manera apropiada. Los subtipos del síndrome congénito se basan en el gen y en el canal afectado (5). En la actualidad se han descubierto más de 300 mutaciones en los cinco genes conocidos que se asocian al SQTL congénito y cada mutación puede tener diferentes implicaciones clínicas. La forma congénita del SQTL se presenta en uno de cada 5.000 nacidos vivos y es una de las principales causas de muerte súbita en adolescentes y adultos jóvenes, presentándose en general por debajo de los 40 años y mayor afectación del sexo femenino. El subtipo 1 (LQT1) se ha encontrado aproximadamente en el 50% de las familias a quienes se ha evaluado el genotipo y es el gen responsable principal de los síndromes de Romano-Ward (formas autonómicas dominantes) y del Jervell y Lange-Nielsen (autonómica recesiva), este último acompañado de sordera y provocado por mutaciones en genes causantes de LQT1 y LQT5; el síndrome de Andersen es una enfermedad congénita muy rara, esporádica o autosómica dominante, caracterizada por múltiples dismorfias y muerte súbita; está causado por mutaciones en el gen KCNJ2, que codifica la subunidad alfa del rectificador de entrada de los canales de potasio (6). El LQT4 no es una verdadera canalopatía, pues afecta proteínas de la membrana celular destinadas a agrupar diversos canales, bombas y transportadores, manifestándose con arritmias ventriculares malignas (7).
SQTL adquirido
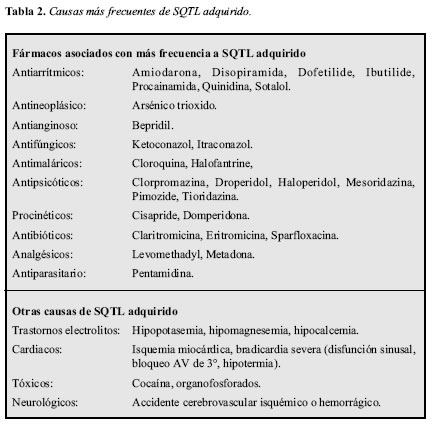
La causa más frecuente de SQTL adquirido es el consumo de algunos medicamentos (8) y se han descrito múltiples situaciones clínicas en las que también se puede presentar. En la Tabla 2 se muestran los medicamentos asociados con mayor frecuencia a prolongación del QT y otras causas no farmacológicas. En la página de internet http://www.qtdrugs.org se puede encontrar un listado más detallado de los medicamentos que pueden prolongar el QT. Existen diferentes condiciones clínicas que pueden facilitar la prolongación del QTc asociado al uso de medicamentos, como la hipertrofia ventricular izquierda, la isquemia miocárdica, la fibrosis miocárdica y alteraciones electrolíticas (9, 10).
Fisiopatología del SQTL adquirido
Las características de las arritmias inducidas por medicamentos son similares a las que se observan en el síndrome congénito y se relacionan con el bloqueo de los canales de potasio (IKr), de la misma forma que ocurre en el subtipo del SQTL congénito tipo 2 (11). En condiciones normales el uso de un medicamento con estas propiedades no se asocia con una prolongación del intervalo QTc, sin embargo, en presencia de una predisposición subclínica, denominada reserva disminuida de la repolarización, el medicamento puede prolongar anormalmente el intervalo QT y facilitar una taquicardia de "puntas torcidas". El potencial de acción en los miocitos está determinado por el balance dinámico entre las corrientes iónicas de entrada y de salida a través de las membranas celulares, de manera que la disminución de las corrientes de salida o el aumento del flujo de iones hacia adentro, pueden retrasar la repolarización y prolongar la duración del potencial de acción (12). La prolongación del potencial de acción en algunas células cardíacas puede resultar en pospotenciales precoces. Las células miocárdicas más susceptibles a la generación de pospotenciales precoces son las células de la red de Purkinje y las células M (células de miocardio intermedio), pues el potencial en estas células tienen una duración mayor, comparado con las células del epicardio y del endocardio (13).
Los pospotenciales precoces pueden ocurrir cuando la repolarización se retrasa lo suficiente durante la fase meseta o fase 2, en donde se puede recuperar una fracción de los canales inactivados de calcio, tipo L, pudiendo volverse a abrir para permitir corrientes de iones hacia adentro, que inducen los pospotenciales precoces. Estos pospotenciales precoces, en algunas situaciones pueden excitar una mayor cantidad de tejido miocárdico repolarizado para alcanzar el umbral y desencadenar otro potencial, y si se propaga a través de los ventrículos, originarán un latido ventricular prematuro. En el SQTL, existe una heterogeneidad exagerada de la duración del potencial de acción a través de los ventrículos, y por tanto la actividad ventricular prematura puede llevar a la excitación de circuitos complejos de reentrada, resultando en la formación de taquicardia ventricular polimórfica. Las arritmias reentrantes polimórficas como consecuencia de bloqueos por la dispersión de la refractariedad y la actividad gatillada mediada por los pospotenciales precoces pueden constituir los mecanismos de las arritmias en el SQTL (14).
Cambios en el intervalo QT relacionado con isquemia miocárdica
Durante la isquemia y la hipoxia miocárdica aguda, se observa una salida de potasio intracelular y su acumulación a nivel extracelular; esta pérdida de potasio es debida en parte al incremento intracelular de sodio, lo cual implica que la bomba sodio/potasio ATPasa es incapaz de compensar la sobrecarga de iones sodio que entran a los miocitos. No está claro si este disbalance iónico resulta de un incremento primario en la recaptación de sodio más allá de la máxima capacidad de la bomba sodio/potasio ATPasa, a la inhibición primaria de la bomba o de ambos. La disminución del contenido intracelular de ATP por la isquemia también puede inhibir la bomba sodio/potasio ATPasa. El flujo hacia afuera de potasio durante la isquemia involucra los canales de potasio dependientes de ATP, los canales de potasio rectificadores con flujo de entrada, a otros canales de potasio dependientes de voltaje y potencialmente también los hemicanales plasmáticos de conexina-43 (15).
El aumento de las concentraciones intracelulares de sodio se asocia a efectos electrofisiológicos deletéreos: resulta en un incremento de la concentración intracelular de calcio, el cual se produce por un incremento del flujo de entrada de calcio a través de la acción reversa de la bomba sodio/calcio, posiblemente por la activación de los canales de calcio tipo L y por el flujo hacia adentro de calcio a traves de hemicanales plasmáticos de conexina-43. La sobrecarga celular de calcio secundaria a la sobrecarga de sodio aumenta la probabilidad de que se induzcan pospotenciales. En las etapas iniciales del infarto de miocardio de menos de 12 horas, el intervalo QT disminuye en promedio 13 ms en la pared inferior y 22 ms en la pared anterior. Después de este tiempo el intervalo QT nuevamente aumenta, en promedio 37 ms en la pared inferior y 52 ms en la pared anterior. El acortamiento inicial pudiera estar explicado, en parte, por los profundos cambios simpáticos y parasimpáticos a nivel cardíaco.
La prolongación del intervalo QT que ocurre en el infarto se relaciona con diferentes factores, entre los cuales están la fracción de eyección del ventrículo izquierdo, la extensión y severidad de la enfermedad coronaria y la presencia de arritmias ventriculares en las primeras 24 horas del infarto (16). Las arritmias ventriculares polimórficas relacionadas con un infarto, que ocurren durante la fase hiperaguda del mismo, se relacionan claramente con la isquemia y no con prolongación del intervalo QTc. Por otra parte, existen otro tipo de arritmias ventriculares polimórficas que se presentan durante la fase de cicatrización del infarto, en pacientes sin evidencia de isquemia sobreagregada y en este caso sí suele haber una prolongación excesiva del intervalo QT (17). Cuando el paciente presenta prolongación anormal del intervalo QT y presenta taquicardia ventricular en "puntas torcidas" dependiente de una pausa, en ausencia de una causa identificable, se puede denominar "SQTL relacionado con infarto de miocardio" que se caracteriza porque: 1) Se observan cambios marcados en la morfologia del intervalo QT después de las pausas. 2) Existe un intervalo de acoplamiento prolongado de las extrasístoles que desencadenan la taquicardia de puntas torcidas (no hay fenómeno de R/T). 3) El potencial de acción de las células de purkinje y células M, que inicialmente se acorta durante la isquemia prolongada, no sólo se normaliza durante la recuperación sino que aumenta; esta prolongación del potencial de acción a menudo induce pospotenciales precoces y a la aparición de arritmias en tejidos infartados. 4) La diferencia en el período refractario entre las áreas adyacentes al infarto es máxima pocos días después del infarto y se normaliza en semanas y 5) Como en otras formas de síndrome de QT prolongado, las "puntas torcidas" después del infarto están precedidas por ciclos corto-largo-corto.
Estas oscilaciones en la longitud de ciclo pueden aumentar la sobrecarga de calcio desencadenando más extrasístoles, mientras que aumenta la dispersión de la refractariedad, básicamente por prolongación del potencial de acción en el miocardio isquémico, facilitando reentradas. La prolongación del intervalo QT en el infarto de miocardio es transitoria, alcanzando el máximo valor a los pocos días y regresando a sus valores iniciales generalmente a los 10 días. Aunque el intervalo QTc generalmente se normaliza, las arritmias ventriculares malignas en "puntas torcidas" pueden ocurrir entre 3 y 10 días despues del infarto. Las ondas T grandes y bizarras que se observan, resultan de un bloqueo combinado de las corrientes IKr e IKs; la denervación simpática irregular y a veces profunda que a menudo sigue al infarto de miocardio, puede contribuir a la prolongación del intervalo QTc y a la arritmogénesis. Para finalizar, la prolongación anormal del QT es infrecuente; dada la relevancia de las consecuencias, debemos tenerla en cuenta cuando prescribimos un fármaco y disponemos de dos poderosas armas: la sospecha clínica y el electrocardiograma.
Referencias
1. Schwartz PJ, Wolf S. QT Interval Prolongation as Predictor of Sudden Death in Patients with Myocardial Infarction. Circulation 1978; 57: 1074 77. [ Links ]
2. Khan IA. Long QT Syndrome: Diagnosis and Management. Am Heart J 2002; 143: 7 14. [ Links ]
3. Priori SG, Aliot C, Blomstrom L, Bossaert L, Breithardt G, Brugada P, Camm J, Cappato R, Cobbe SM, Di Mario C et al. Task Force on Sudden Cardiac Death of the European Society of Cardiology. European Heart Journal 2001; 22: 13741450. [ Links ]
4. Wilde AAM, Bezzina CR. Genetics of Cardiac Arrhythmias. Heart 2005; 91: 135258. [ Links ]
5. Roden DN, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest 2005; 15: 202532. [ Links ]
6. Davies NP, Imbrici P, Fialho D, Herd C, Bilsland LG, Weber A, Mueller R, Hilton-Jones D, Ealing J, Boothman BR, Giunti P, Parsons LM, Thomas M, Manzur AY, Jurkat-Rott K, Lehmann-Horn F, Chinnery PF, Rose M, Kullmann DM, Hanna MG. Andersen-Tawil syndrome: new potassium channel mutations and possible phenotypic variation. Neurology 2005; 65:1083-9. [ Links ]
7. Sherman J, Tester DJ, Ackerman MJ. Targeted mutational analysis of ankyrin-B in 541 consecutive, unrelated patients referred for long QT syndrome genetic testing and 200 healthy subjects. Heart Rhythm 2005; 2:1218- [ Links ]23.
8. Ackerman MJ. Molecular Basis of Congenital and Acquired Long QT Syndromes. J of Electrocardiol 2004; 37 : 1-6. [ Links ]
9. Khan IA. Clinical and therapeutic aspects of congenital and acquired long QT Syndrome. Am J Med 2002; 112:5866. [ Links ]
10. Camm AJ, Janse MJ, Roden DM, Rosen MR, Cinca J, Cobbe SM. Congenital and acquired long QT syndrome. European Heart Journal 2000; 21: 123237. [ Links ]
11. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A Structural Basis for Drug-induced Long QT Syndrome. Proc Natl Acad Sci 2000; 97: 12329-33. [ Links ]
12. Sporton SC, Taggart P, Sutton PM, Walker JM, Hardman SM. Acute ischaemia: a dynamic influence on QT dispersion. Lancet 1997; 349: 3069. [ Links ]
13. El-Sherif N, Caref EB, Yin H, Restivo M. The electrophysiological mechanism of ventricular arrhythmias in the long QT syndrome: tridimensional mapping of activation and recovery patterns. Circ Research 1996; 79: 474-92. [ Links ]
14. Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest 2005; 115: 2305-15. [ Links ]
15. Davey P. QT Interval and mortality from coronary artery disease. Progress in Cardiovasc Dis 2000; 42: 359 84. [ Links ]
16. Halkin A, Roth A, Lurie Ido, Fish R, Belhassen B, Viskin S. Pause-Dependent Torsade de Pointes Following Acute Myocardial Infarction: A Variant of the Acquired Long QT Syndrome. JACC 2001; 38: 116874. [ Links ]

