










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.31 no.3 Bogotá July./Sept. 2006
Proteína reguladora del TNF-a de importancia patogénica en artritis reumatoide
TNF-a regulating protein, of pathogenic importance in rheumatoid arthritis
Dr. Carlos Alberto Cañas Dávila: Internista Reumatólogo, Fundación Valle del Lili, Coordinador Posgrado de Medicina Interna CES-FVL, Cali;
Dr. Antonio Iglesias Gamarra: Internista Reumatólogo, Profesor Titular, Unidad de Reumatología, Departamento de Medicina Interna, Facultad de Medicina, Universidad Nacional de Colombia, Bogotá, D.C.
Correspondencia: Dr. Carlos Alberto Cañas Dávila. Fundación Valle del Lili, Autopista Simón Bolívar, Cra 98 No. 18-49, Tel. (2)3317474 Ext. 7421, Cali, Colombia E-mail: ccanas@telesat.com.co
Recibido: 03/03/06 Aprobado: 30/08/06
Resumen
La tristetraprolina (TTP) hace parte de una familia de proteínas del tipo dedo de zinc con secuencias repetidas CCCH, y consta de tres miembros conocidos en mamíferos, y un cuarto de reciente descubrimiento en ranas y peces. Aunque la TTP fue clonada hace más de 15 años, e identificada como un factor de transcripción, su verdadera función en la síntesis proteica va siendo dilucidada en los últimos años. Se sabe, por ejemplo, que la TTP se une a elementos ricos en AU (adenina-uridina) (AREs) del mRNAs que encoda el factor de necrosis tumoral a (TNF-a) y el factor estimulante de colonias de granulocitos/ macrófagos (GM-CSF). En ambos casos la unión produce desestabilización del mRNA y disminución de la producción de la proteína. Recientemente se tiene evidencia que la TTP puede acelerar la degradación del mRNA por remoción de su cola poliadenilada (deadenilación). En ratones con deficiencia de TTP se desarrolla un síndrome inflamatorio severo con artritis erosiva, autoinmunidad e hiperplasia mieloide. En pacientes con artritis reumatoidea (AR) hay una baja relación TTP/TNF-a indicando una falla en la producción de TTP en respuesta a la producción de TNF-a. Una inapropiada producción de TTP puede ser uno de los factores que contribuye al aumento en la actividad de la AR. El entendimiento de la regulación postranscripcional en la síntesis del TNF-a, es importante para el desarrollo de nuevas estrategias terapéuticas en AR y otras enfermedades en donde el TNF-a tenga importancia patogénica. (Acta Med Colomb 2006; 31: 113-119)
Palabras clave: tristetraprolina, factor de necrosis tumoral-a, artritis reumatoide
Abstract
The tristetraprolin (TTP) family of CCCH tandem zinc-finger proteins is composed of three known members in mammalians, with a fourth member recently identified in frogs and fish. Although TTP was first cloned more than 15 years ago as a transcriptional factor, it is only in the past few years that the physiological function for the protein has been discovered. TTP is now known to bind to the so-called class II AU-rich elements within the mRNAs that encode Tumour Necrosis Factor-a (TNF-a) and granulocyte/macrophage colony-stimulating factor (GM-CSF). In both cases, this binding results in destabilization of the mRNA and decreased secretion of the protein. Recent evidence suggests that TTP can accomplish this accelerated mRNA degradation by first promoting removal of the polyadenylated tail from the mRNA (deadenylation). TTP deficient mice develop a deep inflammatory syndrome with erosive arthritis, autoimmunity and myeloid hyperplasia. In patients with rheumatoid arthritis a low TTP/TNF-a gene expression ratio could indicate failure to produce adequate amounts of TTP in response to increased TNF-a production. Inappropriate TTP production may be one factor that contributes to higher disease activity. Understanding the posttranscriptional regulation of TNF-a biosynthesis is important for the development of new treatment strategies in rheumatoid arthritis and other diseases which TNF-a has pathogenic importance. (Acta Med Colomb 2006; 31: 113-119)
Key words: tristetraprolin, tumour necrosis factor-a, rheumatoid arthritis
Introducción
El factor de necrosis tumoral a (TNF-a) es un mediador de la respuesta inflamatoria aguda y crónica en varias enfermedades (1). Además, ha sido bien conocido su papel en el choque séptico (2-4) y en la caquexia que acompaña al cáncer (5) y al síndrome de Inmunodeficiencia adquirida (6). Los anticuerpos que neutralizan el TNF-a (infliximab, adalimumab) y la proteína quimérica de fusión del receptor p75 (p75-Fc o etanercept), han demostrado ser eficaces en el control de diferentes enfermedades donde el TNF-a es de importancia patogénica, como es el caso de la artritis reumatoide (AR) (7-9). La importancia del TNF-a y sus receptores, han motivado investigaciones en diferentes tópicos que han ampliado bastante su conocimiento en la última década (10, 11). Uno de los tópicos que ha despertado bastante interés es el de los reguladores de la transcripción del TNF-a. Los principales son el TIA-1 (T-cell-restricted Intracellular Antigen) (12), y la TTP (tristetraprolin). Ambos son proteínas que se unen a elementos ricos en "adenina-uridina" (adenine-uridine-rich element o ARE). El TIA-1 inhibe la translación del TNF-a, mientras la TTP promueve la degradación de los transcriptos de TNF-a (13).
La TTP ejerce una función regulatoria del TNF-a, de importancia en la patogénesis de varias enfermedades, lo que hace presumir su papel en el desarrollo de medicamentos para el control de dichas enfermedades (14).
La importancia de la TTP en la patogénesis de la AR y de otras enfermedades similares donde la sobreexpresión del TNF-a tiene importancia en su fisiopatología, hace necesaria la pronta actualización en los médicos que tratan o investigan este tipo de pacientes, situación que motivó para la realización de la presente revisión.
Material y métodos
Realizamos una búsqueda en la base de datos Medline, en lo que respecta a investigaciones publicadas sobre la TTP, haciendo énfasis en sus aspectos moleculares y celulares, su papel en el control de la síntesis del TNF-a, el síndrome experimental por su deficiencia, la TTP humana y las implicaciones en la patogénesis de la AR. Se estudiaron 149 resúmenes, encontrando que 55 de ellos informaban los tópicos objetivo de nuestra revisión, procediendo a la consecución posterior de los artículos originales. Luego de realizar su lectura, encontramos de utilidad ampliar la bibliografía con algunos de los artículos referenciados en dichos trabajos. Es de anotar que la totalidad de los trabajos estudiados fueron investigaciones originales. No se encontraron artículos de revisión actualizados ni en lengua inglesa, ni en castellano. Procedimos a su lectura detallada, análisis y ordenamiento, para la posterior elaboración de la revisión. Fue necesario acudir a algunas ayudas extras en libros de inmunología, biología molecular y biología celular, para complementar la actualización.
Aspectos moleculares y celulares de la TTP
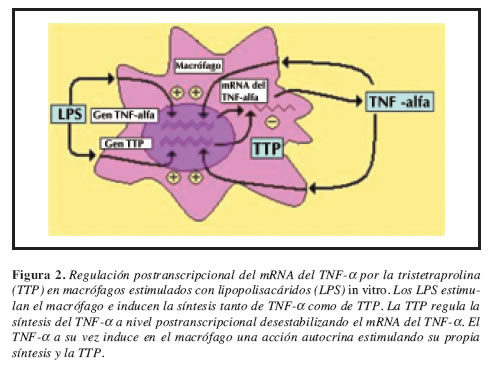
La TTP es el prototipo de una clase de proteínas con configuración en "dedo de zinc" (Figura 1), con dos secuencias distintivas CCCH (Cisteína-Cisteína-Cisteína-Cisteína-Histidina), la cual inhibe la producción del TNF-a, en el macrófago al desestabilizar su RNA mensajero (mRNA). La TTP es una proteína citosólica del macrófago, cuya biosíntesis es inducida por los mismos agentes que estimulan la producción del TNF-a incluyendo el mismo TNF-a (15). Estos hallazgos identifican a la TTP el asa negativa de un mecanismo de autorregulación ("feedback" negativo), que interfiere con la producción del TNF-a, al desestabilizar su mRNA (Figura 2) (16).
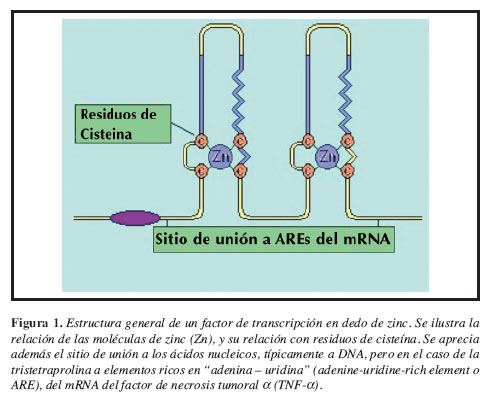
La TTP en animales
La TTP del ratón es una proteína básica, rica en prolina de peso molecular de 33,600 Daltons, contiene tres motivos PPPPG, de allí el nombre de "tetraprolina" (17). Ha recibido también los nombres de Nup475 y TIS11 (18). Como ya se comentó contiene dos dedos de zinc con secuencias CCCH. El gene de la TTP del ratón fue localizado en el cromosoma 7 (19). Recientemente, se han reconocido otras proteínas que contienen estructuras similares como son las encodadas por los genes cDNAs TIS11b (cMG1) en mamíferos (20), TIS11d y DTIS1121 en la Drosophila (22), y sCTH1 y CTH2 de levaduras. Aunque la TTP ha sido localizada en el núcleo celular de fibroblastos de ratón, y debería cumplir funciones como factor de transcripción, no se ha podido hasta el momento encontrar una secuencia en la cual se una al DNA, a diferencia de otras proteínas similares con motivos en dedos de zinc que si lo tienen, y cumplen el papel clásico de factor de transcripción. La TTP cumple su función regulatoria uniéndose al mRNA (23). La expresión del gen que encodan la TTP, el Zfp-36 (Zinc finger protein-36) (24), es inducida por varios mitógenos incluyendo suero, insulina, el factor de crecimiento derivado de las plaquetas (platelet-derived growth factor - PDGF), el factor de crecimiento de fibroblastos (fibroblast growth factor - FGF), y el PMA (phorbol 12-myristate 13-acetate) (14). En el fibroblasto quiescente de ratón, el mRNA del TTP es virtualmente indetectable, pero se acumula en 10 minutos con el estímulo mitogénico, con pico en 30 minutos, retornando a los niveles basales en dos horas. Esto es debido primariamente al ciclo de la transcripción, y que el mRNA tiene una vida media de 15-30 minutos (25). En el ratón, el mRNA es altamente expresado en el pulmón, intestino, nódulos linfáticos bazo y timo, con baja expresión en el hígado y el riñón. En el cerebro la expresión es mínima. Tiene también una alta localización en varias células hematopoyéticas, en macrófagos y en neutrófilos, con baja presencia en linfocitos B y T (1).
El gen que encoda la TTP, Zfp-36, es rápidamente inducido por los factores mitógenos y crecimiento anotados. 77 pares de bases 5' del sitio donde se inicia la transcripción son suficientes para inducir el promotor del Zfp-36. Esta región del promotor incluye secuencias que son sitios de unión para los factores de transcripción EGR-1, AP2 y Sp1 (26). Se ha identificado además un elemento adicional que se une al grupo de proteínas en el sitio del promotor, el cual se ha denominado, "elemento para el promotor del TTP" (TTP promoter element 1 o TPE1); el cual posee una gran similitud en diferentes especies, humana, bovina o murina (27).
Se conoce además que en la TTP como fosfoproteína que es, se llevan a cabo procesos de fosforilación que se estimulan o se inhiben por diferentes estímulos. El PDGF, FGF y el PMA incrementan la fosforilación, más no el cAMP dibutiril o fosfocolina. El residuo de serina 220 es uno de los sitios de fosforilación (28). Dicha fosforilación es mediada por la vía de las quinasas reguladoras p38 MAPK (mitogen-activated protein kinase) (29).
El tratamiento de macrófagos con inhibidores piridinil imidazol de las proteínas quinasa p38 inhibe el estímulo de lipopolisacáridos (LPS) para la secreción del TNF-a. Este mismo procedimiento realizado en macrófagos de la médula ósea de ratones deficientes en TTP fueron menos sensibles al efecto de los inhibidores de p38, a pesar de una respuesta de activación normal con LPS.
La fosforilación de TTP fue incrementada en macrófagos intactos después del estímulo con LPS. Este efecto fue bloqueado por inhibidores p38. Estos resultados hacen sugerir que el TTP es un componente de una cascada de señalización, iniciada por un estímulo proinflamatorio y mediado en parte por la activación de p38, que en últimas lleva a un incremento de la secreción de TNF-a (Figura 3) (30).

Síndrome por deficiencia experimental de TTP
Recientemente, la disrupción del Zfp-36 en células primordiales embrionarias (stem cells) de ratones se ha usado para crear animales que no expresan TTP (31). Estos ratones son normales al nacimiento, pero luego, su crecimiento se frena, y se tornan caquécticos, desarrollan artritis erosiva, conjuntivitis, proliferación mesangial renal, alopecia en parches, dermatitis y altos títulos de anticuerpos antinucleares. A menudo mueren en pocos meses. Además, estos animales tienen hiperplasia mieloide, granulocitosis en bazo, nódulos linfáticos y sangre periférica, disminución de los linfocitos B y T a nivel del bazo y sangre periférica. Este fenotipo sugiere que el TTP realiza un papel en la granulopoyesis o en el desarrollo de los linajes hematopoyéticos. Los análisis de macrófagos derivados de ratones knockout de TTP revelan un incremento de cinco veces en la secreción de TNF-a, y dos veces en los niveles de mRNA del TNF-a. Este incremento de la biosíntesis del TNF-a es debida al incremento de la estabilidad del mRNA. Si a estos modelos animales con carencia de TTP, que semejan modelos animales con exceso de TNF-a (32), se les trata con anticuerpos anti-TNF-a, muchos de los problemas anotados no se desarrollan o se revierten (33), experimento que ayuda a comprender la importancia patogénica de la sobre-expresión de TNF-a. El síndrome de deficiencia de TTP se desarrolla por sobreexpresión de TNF-a en los macrófagos y no en los linfocitos, tanto en forma basal como por estímulo con LPS, como se demostró en estudios celulares in vivo de estos modelos (34).
El papel de los receptores del TNF-a en el desarrollo del síndrome por carencia de TTP se investigó al crear ratones con deficiencia combinada en TTP y TNFR1, en los cuales no se presentó el síndrome, concluyendo que la presencia del receptor es importante para desarrollarse el efecto de la sobreexpresión del TNF-a (35).
La deficiencia de TTP también produce un incremento en la producción celular de GM-CSF (granulocyte-macrophage colony-stimulating factor), e incrementa la estabilidad de su mRNA (36). El TTP parece ser un regulador fisiológico del mRNA del GM-CSF. Estudios preliminares también implican a la TTP como molécula reguladora del mRNA de la interleuquina-3 (37), la interleuquina-2 (38) y c-fos (39).
La TTP se creyó inicialmente que era exclusiva de mamíferos, pero recientemente también ha sido informado en ranas (40) y peces (41).
La TTP humana
La TTP humana, comparte 87% de las secuencias de aminoácidos de la del ratón, y la estructura en dedo de zinc se conserva. El gene humano que encoda la TTP fue localizado en el cromosoma 19 en la banda q13.1 (42). La secuencia de nucleótidos fue informada y confirmada hacia los años noventa por diferentes grupos (43, 44).
Se han explorado polimorfismos genéticos de la TTP humana basados en el estudio de la secuencia genómica del gen Zfp36 y de los dos compartidos o relacionados con otros de mamíferos, el Zfp36L1 y el Zfp36L2, en diferentes regiones geográfica y etnias. Se han identificado 13 polimorfismos en las regiones que codifican genes de las tres proteínas. Una de las mutaciones del Zfp36L1 previene el proceso de splicing de un intrón. Esta mutación fue identificada en un solo alelo de una secuencia de 144 en dos mujeres nativas de Africa Central. El análisis del mRNA en los linfocitos de uno de estos sujetos, confirma que los niveles de mRNA de Zfp36L1 fueron aproximadamente el 50%, comparados con sujetos sin la mutación (45). La significancia funcional de este y otros polimorfismos falta por determinar. Futuras investigaciones están encaminadas a reconocer nuevos polimorfismos y su posible relación con enfermedades con evidencia de excesiva acción del TNF-a.
Brook SA et al. (46) desarrollaron un modelo para caracterizar la unión del TTP humano en la región 3'-UTR (región no translocada) del mRNA del TNF-a, utilizando técnicas de transfección del gen, reacción de cadena de la polimerasa con transcriptasa reversa e inmunohistoquímica. Este grupo también evaluó la distribución del TTP humano a nivel subcelular utilizando un anticuerpo monoclonal específico. El TTP humano interactúa con el mRNA del TNF-a en el citoplasma, en donde la presencia del 3'-UTR del mTNF-a fue necesaria para la unión del TTP humano in vivo. También encontraron que tanto el TTP humano endógeno o transfectado se localiza en el citoplasma. Con técnicas de fraccionamiento de densidad de sucrosa, se evidenció su acumulación en patrones vesiculares en el citosol (47).
Control de la síntesis del TNF-a por la TTP
El TNF-a es expresado por los linfocitos y macrófagos, siendo crítico su papel en los procesos inflamatorios de enfermedades como la AR (48). La activación de los macrófagos produce un incremento de 10.000 veces en la biosíntesis del TNF-a con solo incrementar tres veces su transcripción (49). Así, la transcripción del TNF-a es primariamente regulada a nivel de la estabilidad y translación del mRNA (50). Esta regulación postranscripcional del TNF-a es mediada a través de elementos ricos en AREs, en su región no translocada (3´UTR) (51). La unión de los AREs y su función parecen requerir la integridad del dominio de unión del dedo de zinc, dado que mutaciones a dicho nivel fallan en desestabilizar el mRNA del TNF-a.
El efecto bioquímico de la TTP parece ser a través de la promoción de la deadenilación de los AREs por interacción con poli(A) ribonucleasa. Esta interacción puede ser responsable de la habilidad de la TTP y miembros de su familia para promover la deadenilación de tales transcriptos en células intactas (52, 53). Es de anotar que uno de los mecanismos que posee el mRNA para su estabilidad, es la cola poli (A) en el extremo 3´. En la medida que esta cola se va perdiendo por efecto de las ribonucleasas, su estabilidad se va perdiendo y se degrada. Así, mRNA que carece de cola poli (A) se degradan rápidamente, y los que tienen cola poli (A) larga tienen mayor longevidad. Se ha identificado además una proteína de enlace poli(A) (PEPA), que ayuda en parte a su estabilización. Cada PEPA "cubre" 30 residuos de adenina. Interesantemente la degradación ocurre en el extremo 5´ por efecto de exonucleasas (Figura 4) (54). Otro mecanismo de regulación y desestabilización del mRNA, son los llamados "microRNAs", los cuales se investigan además como oncogenes o supresores de tumores (55), y cuya descripción escapa de esta revisión.

Una manera indirecta de control postranscripcional del TNF-a mediada por la TTP es a través de citoquinas que estimulan su síntesis y expresión, como es el caso de la interleuquina-4 a nivel de los mastocitos (56), o del TGF-b (Transforming Growth Factor-b) en los linfocitos T (57). Estas dos citoquinas son bien reconocidas como reguladoras de la respuesta inflamatoria. La interleuquina 10 es inhibitoria del TNF-a, por mecanismo independiente de la TTP (58).
Papel de la TTP en apoptosis
La TTP y las proteínas relacionadas TIS11b y TIS11d, en niveles fisiológicos causan muerte celular por apoptosis en una forma similar a la observada en ciertas oncoproteínas. La TTP pero no la TIS11b o la TIS11d sensibiliza a las células para la inducción de apoptosis mediada por el TNF-a. Los datos sugieren que la TTP y las TIS11b y d son similares bioquímicamente pero tiene distintos efectos biológicos en vías de crecimiento o supervivencia celular. La TTP puede influenciar la regulación del TNF-a en su función proapoptótica (59).
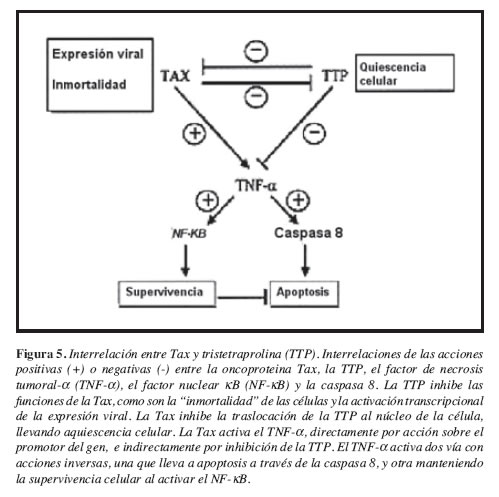
En otras condiciones donde se alteran los mecanismos apoptóticos como en las neoplasias, el papel de la TTP empieza a investigarse. En las leucemias inducidas por retrovirus como la ocasionada por el PTLV (primate T-cell leukemia viruses), la sobreexpresión del TNF-a es importante en su patogenia. La oncoproteína Tax, es un regulador transcripcional de la expresión viral. La TTP tiene una interacción funcional con la Tax, situación que fue investigada en diferentes células de mamíferos (HeLa, D17, HEK 293, and RAW 264.7), transfectadas con la combinación de Tax y TTP. Se obtuvo que la coexpresión de la Tax y la TTP fue asociada con la acumulación de TTP. TTP es una proteína temprana que inhibe la expresión del TNF-a a nivel posttranscripcional, y la expresión del Tax revierte esta inhibición tanto en macrófagos transfectados como en líneas de macrófagos intactos, lo que conduce a la sobre-expresión del TNF-a y su papel mediador para el desarrollo de la neoplasia (60). Las interacciones moleculares de la Tax y la TTP se ilustran en la Figura 5.
Implicaciones en AR
El entendimiento de los mecanismos de regulación pos-transcripcionales del TNF-a es de gran importancia para el desarrollo de estrategias terapéuticas para la AR. La TTP desempeña un papel central en este control, tanto que en modelos animales carentes de esta proteína reguladora se desencadena una patología por sobreexpresión del TNF-a con componentes inflamatorios locales y sistémicos similares a la AR. Como se anotó, modelos en animales han demostrado que alteraciones del TTP inducidas experimentalmente, las cuales alteren su función llevan a un aumento de síntesis del TNF-a y generación de enfermedad inflamatoria. Faltaría por investigar si existen en el humano polimorfismos genéticos de la TTP que lleven a alteraciones en los sitios de unión al mRNA del TNF-a, generándose una disregulación de este último con su consecuente sobreexpresión y generación de enfermedad inflamatoria del tipo AR. El TTP empieza a ser estudiado a nivel del sinovium reumatoide, encontrándose restringido a las células sinoviales, llegándose a postular que una regulación postranscripcional de la síntesis del TNF-a podría ser útil terapéuticamente en AR.
Tsutsumi et al (61) estudiaron las expresiones génicas del mRNA de la TTP y el TNF-a a través de su medición cuantitativa en el tejido sinovial de pacientes con AR y osteoartritis. Encontraron que la relación TTP/TNF-a se encuentra en relación inversamente proporcional con los niveles de proteína C reactiva y de la velocidad de sedimentación globular. La expresión de TTP, fue menor a la del TNF-a, lo que habla a favor de un mecanismo compensatorio que no fue lo suficientemente bueno para reducir la acción del TNF-a. También ha sido estudiada la TTP en células mononucleadas periféricas de pacientes con AR, y se demuestra que hay aumento de su producción principalmente en fases tempranas de la enfermedad, demostrándose una sobreexpresión no regulada de TNF-a (62). Nuevamente se concluye que una producción inapropiada de TTP puede ser uno de los factores que contribuye a una actividad mayor en la AR tanto a nivel sinovial como periférico.
Las alteraciones de la relación TTP/TNF-a en las células mononucleares periféricas se corrigen con el control terapéutico de la enfermedad (63).
Controlar la expresión transcripcional de una proteína con efecto patogénico es uno de los motivos de investigaciones actuales con el fin de buscar medicamentos biológicos que generen desestabilización del mRNA (64), siendo la TTP uno de las posibles candidatos.
Referencias
1. Kollias G, Douni E, Kassiotis G, Kontoyiannis D. The function of tumour necrosis factor and receptors in models of multi-organ inflammation, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Ann Rheum Dis 1999; 58 (Supl 1): 132-9. [ Links ]
2. Tracey KJ, Lowry SF, Fahey TJ III. Cachectin/tumor necrosis factor induces lethal shock and stress hormone responses in the dog. Surg Gynecol Obst 1987; 164:415-22. [ Links ]
3. Bauss F, Droge W, Manuel DN. Tumor necrosis factor mediates endotoxic effects in mice. Infect Immun 1987; 55:1622-5. [ Links ]
4. De Freitas I, Fernandez-Samoza M, Essenfeld-Sekler E, Cardier JE. Serum levels of the apoptosis-associated molecules, Tumor Necrosis Factor-a/Tumor Necrosis Factor type-I Receptor and Fas/FasL in sepsis. Chest 2004;125:2238-46. [ Links ]
5. Bower JE, Ganz PA, Aziz N, Fahey JL. Fatigue and proinflammatory cytokine activity in breast cancer survivors. Psych Med 2002; 64:604-11. [ Links ]
6. Aukrust P, Liabakk NB, Muller F, Lien E, Espevik T, Froland SS. Serum levels of tumor necrosis factor-alpha (TNF alpha) and soluble TNF receptors in human immunodeficiency virus type 1 infection - correlations to clinical, immunologic, and virologic parameters. J Infect Dis 1994;169: 420-4. [ Links ]
7. Klareskog L, van der Heijden, de Jager JP, Gough A, Kalden J, Malaise M. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double-blind randomised controlled trial. Lancet 2004; 363: 675-81. [ Links ]
8. Maini R, St Clair EW, Breedveld F, Furst D, Kalden J, Weisman M. Infliximab (chimeric anti -tumor necrosis factor a monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. Lancet 1999; 354: 1932-9. [ Links ]
9. Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA. Adalimumab, a fully human anti-tumor necrosis factor alpha monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum 2003; 48:35-45. [ Links ]
10. MacEwan DJ. TNF ligands and receptors a matter of life and death . Br J Pharmacol 2002; 135: 855-75. [ Links ]
11. Galarza C, Pineda-Tamayo R, Zurita L, García-Carrasco M, Rojas, J, Anaya JM. Terapia anti-TNF: bases moleculares y utilidad clínica. En: JM Anaya, Shoenfeld Y, Correa PA, García-Carrasco M, Cervera R (Editores). Autoinmunidad y enfermedad autoinmune. Corporación para Investigaciones Biológicas (CIB). Medellín (Colombia); 2005: 432-9. [ Links ]
12. Piecyk M, Wax S, Beck ARP, Kedersha N, Gupta M, Maritim B. TIA-1 is a translational silencer that selectively regulates the expression of TNF-a. EMBO J 2000; 19: 4154-63. [ Links ]
13. Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor a, cyclooxygenase 2, and inflammatory arthritis. Proc Natl Acad Sci U S A 2004; 17: 201116. [ Links ]
14. Tsutsumi A, Suzuki E, Adachi Y, Murata H, Goto D, Kojo S. Expression of tristetraprolin (G0S24) mRNA, a regulator of tumor necrosis factor-alpha production, in synovial tissues of patients with rheumatoid arthritis. J Rheumatol 2004; 31:1044-9. [ Links ]
15. Fairhurst AM, Connolly JE, Hintz KA, Goulding NJ, Rassias AJ, Yeager MP, et al. Regulation and localization of endogenous human tristetraprolin. Arthritis Res Ther 2003; 5: 21425. [ Links ]
16. Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-a production by tristetraprolin. Science 1998; 281:1001-5. [ Links ]
17. Lai WS, Stumpo DJ, Blackshear PJ. Rapid insulin-stimulated accumulation of an mRNA encoding a proline rich protein. J Biol Chem 1990; 265: 16556-63. [ Links ]
18. DuBois RN, McLane MW, Ryder K, Lau L F, Nathans D. A growth factor-inducible nuclear protein with a novel cystein/histidine repetitive sequence. J Biol Chem 1990; 265: 19185-91. [ Links ]
19. Saudenrs AM, Seldin MF. The syntenic relationship of proximal mouse chro mosome 7 and the myotonic dystrophy gene region on human chromosome 19q. Genomics 1990; 6: 324-32. [ Links ]
20. Gomperts M, Pascall JC, Brown K D. The nucleotide sequence of a cDNA encoding an EGF-inducible gene indicates the existence of a new family of mitogen-induced genes. Oncogene 1990; 5: 1081-83. [ Links ]
21. Ma Q, Wadleigh D, Chi T, Herschman H. The Drosophila TIS11 homologue encodes a developmentally controlled gene. Oncogene 1994; 9: 3329-34. [ Links ]
22. Mohler J, Weiss N, Murli S, Mohammadi S, Vani K, Vasilakis g, et al. The embryonically active gene, unkempt, of Drosophila encodes a Cys3His finger protein. Genetics 1992; 131: 377-88. [ Links ]
23. Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor a mRNA. Mol Cell Biol 1999; 19:4311-23. [ Links ]
24. Taylor GA, Lai WS, Oakey RJ, Seldin MF, Shows TB, Eddy RL Jr, et al. The human TTP protein: sequence, alignment with related proteins, and chromosomal localization of mouse and human genes. Nucleic Acids Res 1991; 19:3454 [ Links ]
25. Taylor GA., Blackshear PJ. Zinc inhibits turnover of labile mRNAs in intact cells. J Cell Phys 1995; 162: 378-87. [ Links ]
26. Lai WS, Thompson MJ, Blackshear PJ. Characteristics of the intron involvement in the mitogen-induced expression of Zfp-36. J Biol Chem 1998; 273: 506-17. [ Links ]
27. Lai WS,Thompson MJ,Taylor GA,Liu Y, Blackshear PJ. Promoter analysis of Zfp-36, the mitogen-inducible gene encoding the zinc finger protein tristetraprolin. J Biol Chem 1995; 270: 25266-72. [ Links ]
28. Taylor GA, Thompson MJ, Lai WS, Blackshear PJ. Phosphorylation of tristetraprolin, a potential zinc finger transcription factor, by mitogen stimulation in intact cells and by mitogen-activated protein kinase in Vitro. J Biol Chem 1995; 270: 13341-47. [ Links ]
29. Mahtani KR, Brook M, Dean JLE, Sully G, Saklatvala J, Clark AR. Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol Cell Biol 2001; 21: 6461-9. [ Links ]
30. Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ. Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the Involvement of tristetraprolin in the p38 Signaling Pathway. J Biol Chem 2001; 276: 42580-7. [ Links ]
31. Taylor GA, Carballo E, Lee DM, Lai WS, Thopson MJ, et al. A pathogenetic role of TNF-a in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 1996; 4:445-54. [ Links ]
32. Schwartz RS. Autoimmunity and autoimmune diseases.En: WE Paul editor. Fundamental Immunology. 3rd ed. Raven Press, New York. 1993; 1033-97. [ Links ]
33. Sheehan KCF, Ruddle NH, Schreiber RD. Generation and characterization of hamster monoclonal antibodies that neutralize murine tumor necrosis factors. J Immunol 1989; 142: 3884-93. [ Links ]
34. Carballo E, Gilkeson GS, Blackshear PJ. Bone Marrow Transplantation Reproduces the Tristetraprolin-Deficiency Syndrome in Recombination Activating Gene (-/-) Mice. Evidence That Monocyte/Macrophage Progenitors May Be Responsible for TNF-a Overproduction. J Clin Invest 1997; 100: 986-95. [ Links ]
35. Carballo E, Blackshear PJ. Roles of tumor necrosis factor-a receptor subtypes in the pathogenesis of the tristetraprolin-deficiency syndrome. Blood 2001; 98: 2389-95. [ Links ]
36. Carballo E, Lai WS, Blackshear PJ. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood 2000; 95: 1891-9. [ Links ]
37. Stoecklin G, Ming XF, Looser R, Moroni C. Somatic mRNA Turnover Mutants Implicate Tristetraprolin in the Interleukin-3 mRNA Degradation Pathway. Mol Cell Biol 2000; 20: 375363. [ Links ]
38. Ogilvie RL, Abelson M, Hau HH, Vlasova I, Blackshear PJ, Bohjanen PR. Tristetraprolin down-regulates IL-2 gene expression through AU-rich element-mediated mRNA decay. J Immunol 2005; 174: 953-61. [ Links ]
39. Raghavan A, Robison RL, McNabb J, Miller CR, Williams DA, Bohjanen PR. HuA and tristetraprolin are induced fllowing T cell activation and display distinct but overlapping RNA-binding specificities. J Biol Chem 2001; 276: 47958-65. [ Links ]
40. De J, Lai WS, Thorn JM, Goldsworthy SM, Liu X, et al. Identification of four CCCH zinc finger proteins in Xenopus, including a novel vertebrate protein with four zinc fingers and severely restricted expression.Gene 1999; 228: 13345. [ Links ]
41. Blackshear PJ, Wilson S, Suk W. Biomarkers of Environmentally Associated Disease: Technologies, Concepts and perspectives. 2 da ed. Boca Raton, FL: Editorial CRC; 2002.p. 33953. [ Links ]
42. Shows TB, Sakaguchi AY, Naylor SL. Mapping the human genome, cloned genes, DNA polymorphisms, and inherited disease. Adv Hum Genet 1982; 12: 341-452. [ Links ]
43. Varnum BC, Lim R W, Sukhatme VP, Herschman HR. Nucleotide sequence of a cDNA encoding TIS11, a message induced in Swiss 3T3 cells by the tumor promoter tetradecanoyl phorbol acetate. Oncogene 1989; 4: 11920. [ Links ]
44. Varnum BC, Ma QF, Chi TH, Fletcher B, Herschman HR. The TIS11 primary response gene is a member of a gene family that encodes proteins with a highly conserved sequence containing an unusual Cys-His repeat. Mol Cell Biol 1991; 11: 1754-8. [ Links ]
45. Blackshear PJ, Phillips RS, Vazquez-Matias J, Mohrenweiser H. Polymorphisms in the genes encoding members of the tristetraprolin family of human tandem CCCH zinc finger proteins. Prog Nucleic Acid Res Mol Biol 2003; 75: 43-68. [ Links ]
46. Brooks SA, Connolly JE, Diegel RJ, Fava RA, Rigby WF. Analysis of the function, expression, and subcellular distribution of human tristetraprolin. Arthritis Rheum 2002; 46: 1362-70. [ Links ]
47. Cao H, Tuttle JS, Blackshear PJ. Immunological characterization of tristetraprolin as a low abundance, inducible, stable cytosolic protein. J Biol Chem 2004; 279: 21489-99. [ Links ]
48. Klippel JH, Dieppe PA. Rheumatology. 2nd ed. Philadelphia: Editorial Mosby; 1998. [ Links ]
49. Beutler B, Cerami A. The biology of cachectin/TNF: primary mediator of the host response. Annu Rev Immunol 1989; 7: 625-55. [ Links ]
50. Kruys V, Thompson P, Beutler B. Extinction of tumor necrosis factor locus and of genes encoding the lipopolysaccharide signaling pathway. J Exp Med 1993;177:1383-90. [ Links ]
51. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 1999; 10: 387-98. [ Links ]
52. Blackshear PJ. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem Soc Trans 2002; 30(Pt 6): 94552. [ Links ]
53. Lai WS, Kennington EA, Blackshear PJ. Tristetraprolin and its family members can promote the cell-free deadenylation of AU-rich element-containing mRNAs by Poly(A) ribonuclease. Mol Cell Biol 2003; 23: 3798812. [ Links ]
54. Karp G. Biología celular y molecular. México: Editorial McGraw-Hill Intera mericana; 2003.p. 529-34. [ Links ]
55. Chen CZ. MicroRNAs as oncogenes and tumor suppressors. N Eng J Med 2005; 353: 1768-71. [ Links ]
56. Suzuki K, Nakajima H, Ikeda K, Maezawa Y, Suto A, et al. IL-4-Stat6 signaling induces tristetraprolin expression and inhibits TNF-alpha production in mast cells. J Exp Med 2003; 198: 1717-27. [ Links ]
57. OgawaK, Chen F, KimYJ, Chen Y. Transcriptional regulation of tristetraprolin by transforming growth factor-b in human T cells. J Biol Chem 2003; 278: 30373-81. [ Links ]
58. Kontoyiannis D, Kotlyarov A, Carballo E, Alexopoulou L, Blackshear PJ, Gaestel M, et al. Interleukin-10 targets p38 MAPK to modulate ARE-dependent TNF mRNA translation and limit intestinal pathology. EMBO J 2001; 20: 3760-70. [ Links ]
59. Johnson BA, Geha M, Blackwell TK. Similar but distinct effects of the tristetraprolin/TIS11 immediate-early proteins on cell survival. Oncogene 2000; 19: 1657-64. [ Links ]
60. Twizere JC, Kruys V, Lefèbvre L, Vanderplasschen A, Collete D, Debacq C, et al. Interaction of retroviral Tax oncoproteins with tristetraprolin and regulation of tumor necrosis factor-a expression. J National Cancer Institute 2003; 95: 1846-59. [ Links ]
61. Tsutsumi A, Suzuki E, Adachi Y, Murata H, Goto D, Kojo S, et al. Expression of tristetraprolin (G0S24) mRNA, a regulator of tumor necrosis factor-alpha production, in synovial tissues of patients with rheumatoid arthritis. J Rheumatol 2004; 31: 1044-9. [ Links ]
62. Fabris M, Tolusso B, Di Poi E, Tomietto P, Sacco S, et al. Mononuclear cell response to lipopolysaccharide in patients with rheumatoid arthritis: relationship with tristetraprolin expression. J Rheumatol 2005; 32: 998-1005. [ Links ]
63. Fabris M, Tolusso B, Gremese E, Tomietto P, Ferraccioli G. Analysis of the kinetic of expression of tristetraprolin and HuR by rheumatoid arthritis patients peripheral blood mononuclear cells stimulated with lipopolysaccharide. Reumatismo 2004; 56:94-103. [ Links ]
64. Williams BR. Targeting specific cell types with silencing RNA. N Eng J Med 2005; 353:1410-11. [ Links ]

