










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.31 no.4 Bogotá Oct./Dec. 2006
Reporte de un caso
Nephrotic syndrome, secondary to lecithin-cholesterol acyltransferase deficiency
Case report
Dr. Gustavo A. Marín: Internista, Nefrólogo Centro Terapia Renal Bolivariana, Clínica Universitaria Bolivariana, Medellín; Dr. José L. Torres: Residente II Medicina Interna Universidad Pontificia Bolivariana; Dra. Rocío López: Patóloga, Servicio de Patología Fundación Santa Fe de Bogotá; Dr. Rafael Andrade: Jefe Departamento Patología y Laboratorios, Fundación Santa Fe de Bogotá, Colombia.
Correspondencia: Dr. José Luis Torres Gonzales. Tel.2656531, Medellín. E-mail: jtorres@une.net.co
Recibido: 26/06/06 Aceptado: 25/10/06
Resumen
La triada de anemia, síndrome nefrótico y opacidad corneal sugiere deficiencia de lecitin-colesterol aciltransferasa (LCAT), desorden familiar autosómico recesivo que cursa con altos niveles de fosfatidilcolina y colesterol no esterificado con formación de una lipoproteína anormal (lipoproteína X). La acumulación de componentes lipídicos ocurre en sitios intra y extracelulares. Reportamos un caso de síndrome nefrótico e insuficiencia renal crónica estadio 3 con opacidad corneal, anemia no hemolítica, colesterol HDL bajo, triglicéridos elevados y biopsia renal con membrana basal de 1666 nm por presencia de estructuras vacuolares lipídicas y espacios lacunares.
Palabras clave: síndrome nefrótico, deficiencia lecitin-colesterol aciltransferasa (LCAT), lipoproteína X.
Abstract
The triad of anemia, nephrotic syndrome and corneal opacity suggests lecithin-cholesterol acyltransferase (LCAT) deficiency, family autosomal recessive disorder that is studied with increased levels of phosphatidylcholine, unesterified cholesterol in the blood and with the formation of an abnormal lipoprotein ( lipoprotein X ). Accumulation of lipid component occurs in both, intracellular and extra cellular sites. We report a case of Nephrotic Syndrome and Chronic renal failure in stage 3 with corneal opacity, non hemolytic anemia, cholesterol HDL reduced, high triglycerides and renal biopsy with basement membranes 1666 NM showing intramembranous lacunae that seem to contain bubbles and have a vacuolated appearance.
Key words: nephrotic syndrome, lecithin-cholesterol acyltransferase (LCAT) deficiency, lipoprotein- X.
Introducción
Gjone y Norum reportaron en 1967 un desorden familiar caracterizado por proteinuria, anemia, hiperlipidemia y opacidad corneal. La mayoría de los casos de esta enfermedad se describieron inicialmente en los países nórdicos, pero se han descrito también casos en otros países. Es una rara enfermedad de carácter autosómico recesivo causada por mutación en el gen que codifica la lecitin-colesterol aciltransferasa (LCAT) localizado en el cromosoma 16q22. La LCAT cataliza la formación de ésteres de colesterol por vía de hidrólisis y transfiere el ácido graso sn-2 de la fosfatidilcolina al grupo 3-hydroxilo de colesterol. Una deficiencia de esta enzima conlleva un incremento de nivel de fosfatidilcolina y colesterol no esterificado en sangre con la consecuente formación de una lipoproteína anormal llamada lipoproteína X. Clínicamente se manifiesta en la tercera o cuarta década de la vida con depósito progresivo de lípidos en varios tejidos incluyendo el riñón, con esclerosis glomerular e inclusive falla renal terminal.
Caso clínico
Paciente masculino de 33 años, quien consultó en octubre de 2004 con edema de dos meses de evolución y síntomas respiratorios con tos, expectoración purulenta y fiebre no cuantificada de 15 días de evolución por lo cual consulta. El paciente no tenía antecedentes personales de importancia, ni familiares. Al examen físico en buen estado general TA 130/95 sentado FC 84x' FR 20x'; con signos de consolidación y derrame pleural basal izquierdo y edemas en miembros inferiores.
Se consideró neumonía adquirida en la comunidad (NAC) con derrame pleural, para lo que recibió manejo antibiótico. Se evidenció síndrome nefrótico y disfunción renal con creatinina (Cr) 1.8 mg/dL, depuración de Cr en orina 24 h de: 54,8 ml/min, volumen urinario (Vol.U.): 2121 cc, Cr urinaria 67 mg/dL, proteinuria de 12,5 gr/24 h, albúmina 2.0 grdL, colesterol 250 mg/dL, triglicéridos (TG) 980 mg/dL. Presentó cifras tensionales ligeramente elevadas. Ingresa con impresión diagnóstica de:
1. Síndrome nefrótico secundario a una glomerulonefritis en estudio
2. Enfermedad renal crónica (IRC) en estadio 3.
3. Neumonía adquirida en comunidad (NAC) con derrame paraneumónico.
Se decidió completar esquema antibiótico y realizar estudios adicionales antes de llevarlo a biopsia renal. Los exámenes paraclínicos de control un mes posterior a su primera visita muestran:
Cr sérica de 1,9 mg/dL, Vol.U. 2200cc/día, depuración de Cr 54 ml/min, Cr urinaria 68 mg/dL, proteinuria de 12 gr/24 hrs. Hemograma con hemoglobina (Hb): 9,5 gr/dL leucocitos 9800 mm3, neutrófilos (N) 74%, linfocitos (L) 18% plaquetas 509.000
Uroanálisis: Ph 7,0, densidad (d) 1005, proteínas 300 mgr/dL
Proteínas séricas 6.1 gr/dL, albúmina 2.5 gr/dL, globulinas 3.6 gr/dL, K 3,9 mmol/L, Na 139 mmol/L, Cl 94 mmol/L.
C3-C4 normales, ANAS negativos, ANTIDNA negativo, ENAS negativos , Ag HBs negativo, anti HVC negativo, HIV negativo, VDRL negativo.
Con estos hallazgos clínicos y de laboratorio y ante la sospecha de una posible glomeruloesclerosis focal y segmentaria como causante del síndrome nefrótico asociado a enfermedad renal crónica grado 3, se decide practicar biopsia renal.
Se realiza biopsia renal bajo ecografía obteniéndose un cilindro que se remite a patología para realizar estudios de microscopía de luz, inmunofluorescencia y microscopía electrónica:
El estudio de microscopía de luz mostró en total 16 glomérulos, cinco de ellos con esclerosis global y tres con glomeruloesclerosis segmentaria. Como hallazgos de importancia se encontró glomerulomegalia moderada y cambios focales por hipoperfusión. Las membranas basales capilares glomerulares mostraron un marcado engrosamiento identificándose dobles contornos y colapso focal. El hallazgo más llamativo fue la presencia de imágenes intraluminales semejantes a los trombos hialinos vistos en nefropatías mediadas por complejos inmunes y la crioglobulinemia y de células de aspecto espumoso en capilares glomerulares (Figuras 1 y 2)
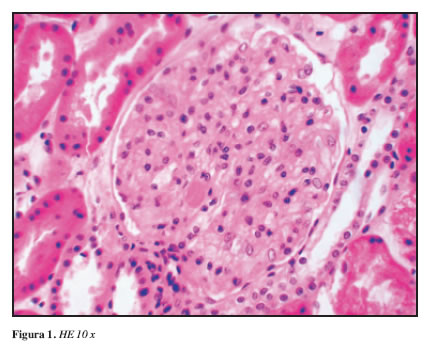
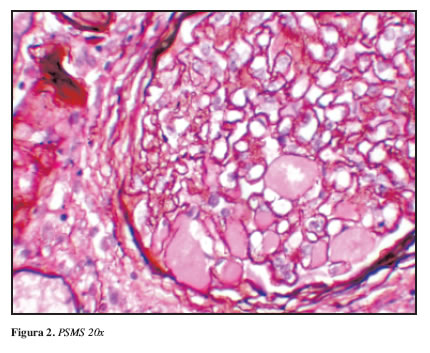
A nivel tubulointersticial, fibrosis del 10% del parénquima y nefritis intersticial con eosinófilos. Los vasos mostraron esclerosis arterial moderada con hialinosis arteriolar. Los hallazgos de la inmunofluorescencia descartaron presencia de complejos inmunes mostrando únicamente depósitos de IgM en áreas de esclerosis segmentaria.
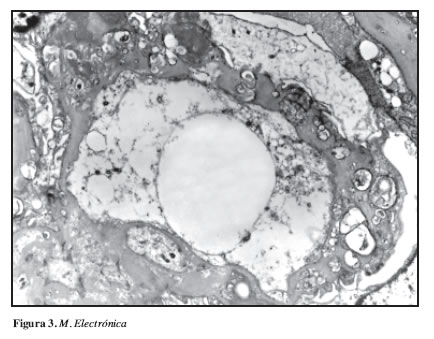
A nivel ultraestructural, los hallazgos más importantes estuvieron centrados en las membranas basales capilares, teniendo un grosor medio de 1666 nm (Valor de referencia 340 ± 70 nm). Este engrosamiento tan marcado estuvo dado por la identificación de estructuras intramembranosas lamelares redondeadas, concéntricas e irregulares, vacuolas lipídicas y espacios lacunares a lo largo de la membrana basal capilar en localización intramembranosa, subendotelial y subepitelial (Figura 3).
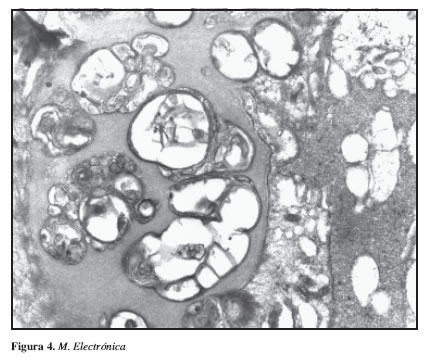
Igualmente se encontraron estas estructuras a nivel mesangial. Se encontró un daño moderado en las células epiteliales viscerales dado por fusión de los procesos de los podocitos y transformación microvellosa. No se identificó presencia de complejos inmunes en ninguna localización. La matriz mesangial se encontró expandida con presencia de las estructuras mencionadas (Figura 4).
Con estos hallazgos se realizó un diagnóstico patológico de una deficiencia de lecitin-colesterol aciltransferasa. (LCTA), una nefritis intersticial crónica en actividad de posible etiología por hipersensibilidad medicamentosa y una esclerosis arterial con hialinosis arteriolar.
Nueve meses después reingresa con un cuadro clínico consistente en toxicodermia medicamentosa con anemia, oliguria y elevación de nitrogenados (creatinina 9 mg/dL, BUN 100 mg/dl) con agudización de falla renal muy probablemente por nefritis tubulointersticial aguda, requiriendo terapia de remplazo renal con hemodiálisis intermitente por dos semanas y esteroides por dos meses, con recuperación de la función renal a su estado previo.
Fue evaluado por hematología por anemia persistente microcítica sin ferropenia. Los estudios mostraron reticulocitos corregidos normales, coombs directo negativo, bilirrubinas normales y ldh en límite superior normal, por lo que se descartó anemia hemolítica autoinmune. El mielograma fue reportado como normal. Electroforesis de Hb normal a solicitud de hematología sin evidencia de rasgo talasémico.
Otros exámenes de laboratorio mostraron: colesterol total 273 mg/dL, HDL 4.0 mg/dL, TG 1260 mg/dL, Cr 1.5 mg/dL, albúmina 3.9 gr/dL, BUN 55 mg/dL, depuración de Cr corregida 41 ml/min, Vol.U. 1750 ml/d, Cr urinaria 50.6 mg/dL, proteinuria 2.67 gr/d, uroanálisis d 1020, pH 5.5, proteínas 300 mg/dl, Erit 5xc, leucocitos 4xc. Hemograma de control con Hb 10.9 gr/dL, VCM 87.8, leucocitos 6310, plaquetas 259000 aglutininas frío negativo, ferritina 1260, K 5.0 meq/l, Ca 8.7 mg/dL, Na 145 meq/l.
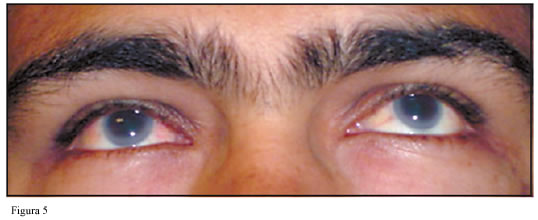
El examen oftalmológico mostró la presencia de opacidad corneal con arco lipoideo (Figura 5). Este hallazgo en conjunto con la documentación de hiperlipidemia con HDL disminuida, anemia normocrómica, proteinuria significativa e insuficiencia renal crónica en estadio 3 es característico de la deficiencia de LCAT.
El paciente continúa con tratamiento médico con inhibidores de enzima convertidora de angiotensina (IECAS), gemfibrozil, carbonato de calcio y furosemida.
Discusión
La deficiencia de la enzima lecitin colesterol-aciltransferasa (LCAT), es una enfermedad heredada como una condición recesiva, comprometiendo ambos sexos. Los niveles plasmáticos se pueden ver afectados por la interacción de factores ambientales, metabólicos y genéticos.
En cuanto a las variables genéticas que codifican los diversos cambios del metabolismo de HDL, encontramos la apolipoproteína A- I (APO A I) principal constituyente proteico de la HDL, las lipasas hepática y lipoproteica, la enzima de transferencia de ésteres de colesterol (CETP), la enzima esterificadora de colesterol libre (LCAT), el transportador ABCA- 1, y el receptor de HDL SR- B1.
Shiro Jimi, Noriko Uesugi y cols (1) presentaron un estudio de cinco pacientes con déficit de LCAT, encontrando mutaciones en cada uno de los casos, unas de características homocigóticas y otras heterocigóticas.
Las diversas mutaciones en los genes de LCAT, son responsables de la actividad de la enzima, y de muchas de la manifestaciones clínicas de la enfermedad (2, 4), sin embargo se considera que otros factores diferentes a la mutación de genes tienen un rol en el desarrollo de esta patología. La LCAT junto con la HDL catalizan la conversión de colesterol no esterificado y fosfatidilcolina a colesterol esterificado y lisofosfatidilcolina (5). La deficiencia de esta enzima causa una acumulación de colesterol no esterificado y al mismo tiempo una disminución de colesterol esterificado por lo cual se presentan finalmente una serie de anormalidades que involucran todas las clases de lipoproteínas y sus estructuras. Tales anormalidades en los lípidos causan alteraciones en los órganos como el riñón, eritrocitos y córnea.
El compromiso de órganos se traduce en enfermedad renal crónica (7, 8), anemia hemolítica y opacidad corneal. En cuanto a la disfunción renal la glomeruloesclerosis es la principal causa de morbimortalidad en el déficit de LCAT familiar (7, 9). Se inicia con una proteinuria progresiva crónica, hematuria y posteriormente enfermedad renal crónica en la cuarta y quinta décadas de la vida, cursando asintomático por décadas y progresando finalmente a falla renal donde su patogénesis no es del todo entendida (11). La biopsia renal muestra la presencia de vacuolización de la membrana basal glomerular, como panal de abejas, con material lamelar, serpiginoso o granular, lesiones llenas de material lipídico y espumoso. Expansión mesangial con células espumosas. El compromiso glomerular se acompaña de daño tubular, infiltrado inflamatorio con atrofia tubular y fibrosis. La microscopía electrónica con estructuras espumosas en mesangio, subendotelio y membrana basal que no son comunes a otras patologías que comprometan el riñón (11-14).
Acumulación extracelular de partículas lipídicas mayores que LDL, o más pesadas que apo- B han sido descritas en glomérulos de pacientes con varias enfermedades glomerulares (14-15). Además fuertes depósitos de LDL oxidada han sido demostrados en lesiones de GMEFyS (glomeruloesclerosis focal y segmentaria) (16). Hyun Soon Lee y Howard S Kruth (17) describen la frecuencia de depósitos de colesterol en 133 pacientes con enfermedad glomerular, 58% de ellos presentaron colesterol esterificado y no esterificado en los glomérulos, con alto porcentaje de esclerosis glomerular y disminución del aclaramiento de creatinina.
La razón de acumulación extracelular de colesterol esterificado en enfermedad glomerular no está clara. Los depósitos de colesterol en las lesiones de GMEFyS como causa o efecto de glomeruloesclerosis es poco claro. Estudios de microscopía electrónica demuestran acumulación de vesículas lipídicas granulares similares a las descritas en casos de déficit LCAT familiar.
Se ha descrito recurrencia de deficiencia LCAT después de trasplante renal y puede reaparecer el daño renal (18).
La anemia hemolítica puede ser explicada por alteraciones en los lípidos de la membrana de las células sanguíneas con la consecuente fragilidad.
Niveles elevados de esfingomielina se han presentado en la córnea de pacientes con déficit de LCAT.
La LCAT afecta los niveles de colesterol HDL plasmáticos. El colesterol libre es removido de los tejidos periféricos pasando por hígado y eliminado del organismo. Al generarse disminución de HDL se aumentan los riesgos de arterioesclerosis. El riesgo aterogénico es variable y los mecanismos que llevan a aterogénesis pobremente entendidos.
Muchos pacientes que presentan esta enfermedad no tienen incrementado el riesgo de sufrir enfermedad coronaria temprana. En adición a la disminución de HDL la deficiencia de LCAT reduce en el plasma los niveles de lipoproteínas proaterogénicas que contienen APO B, por aumento de la expresión hepática de LDL y APO E. Dentro de los fenotipos observados se incluye un incremento en el colesterol libre, colesterol esterificado, hipertrigliceridemia, LDL y VLDL morfológicamente anormales y vesículas ricas en fosfolípidos conocidas como lipoproteínas X ( LP X ).
Una dieta baja en lípidos y medicamentos hipolipemiantes no han mostrado ser beneficiosos.
Conclusión
Reportamos entonces el caso de un paciente con síndrome nefrótico, enfermedad renal crónica estadio 3 secundaria a déficit LCAT- nefritis tubulointersticial crónica activa (NTIC) de posible origen por hipersensibilidad, con anemia no hemolítica, triglicéridos elevados, colesterol HDL bajo, opacidad corneal, y con hallazgos histológicos característicos en la biopsia renal. La enfermedad renal crónica generalmente ocurre en la cuarta década de la vida siendo la mayor causa de morbilidad y mortalidad en esos pacientes. Se resalta por tanto la importancia de un cuidadoso examen físico de los pacientes con enfermedad renal inexplicada, enfocando la atención a múltiples órganos involucrados que puedan sugerir el diagnóstico y la necesidad en todos estos pacientes de realizar una biopsia renal que incluya estudios completos de microscopía de luz, inmunofluorescencia y ultraestructura.
Referencias
1. Shiro Jimi, Noriko Uesugi and Cols. Possible induction of renal dysfunction in patients with Lecithin: cholesterol acyltranferase deficiency by oxidized phosphatidylcholine in glomeruli. Arterioscler Thromb Vasc Biol 1999 ;19: 794801 [ Links ]
2. Frasca GM, Soverini L, Tampieri E, Franceschini G, Calabresi L, Pisciotta L, et al. A 33-year-old man with nephrotic syndrome and lecithin-cholesterol acyltransferase (LCAT) deficiency. Description of two new mutations in the LCAT gene. Nephrol Dial Transplant 2004; 19: 162224 [ Links ]
3. Lynn EG, Siow YL, Frohlich J, Cheung GT, O K. Lipoprotein-X stimulates monocyte chemoattractant protein-1 expression in mesangial cells via nuclear factor-kappa b. Kidney Int 2001; 60: 52032 [ Links ]
4. Funke H, Von Eckardstein A, Pritchard PH, Hornby AE, Wiebusch H, Motti C. Genetic and phenotypic heterogeneity in familial lecithin: cholesterol acyltransferase (LCAT) deficiency. Six newly identified defective alleles further contribute to the structural heterogeneity in this disease. J clin Invest 1993; 91: 67783 [ Links ]
5. Kuivenhoven JA, Pritchard H, Hill J, Frohlich J, Assmann G, Kastelein J. The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J Lipid Res 1997; 38: 191205 [ Links ]
6. Nq DS, Maquire GF, Wylie J, Ravandi A, Xuan W, Ahmed Z, et al. Oxidative stress is markedly elevated in lecithin:cholesterol acyltransferase-deficient mice and is paradoxically reversed in the apolipoprotein E knockout background in association with a reduction in atherosclerosis. J Biol Chem 2002; 277: 1171520 [ Links ]
7. Lambert G, Sakai N, Vaisman BL, Neufeld EB, Marteyn B, Chan CC, et al. Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J Biol Chem 2001; 276: 150908 [ Links ]
8. Zhu X, Herzenberg AM, Eskandarian M, Maguirre GF, Scholey JW, Connelly PW, et al. A novel in vivo lecithin-cholesterol acyltransferase (LCAT)-deficient mouse expressing predominantly LpX is associated with spontaneous glomerulopathy. Am J Pathol 2004; 165: 126978 [ Links ]
9. Imbasciati E, Paties C, Scarpioni L, Mihatsch. Renal lesions in familial lecithin-cholesterol acyltransferase deficiency. Ultrastructural heterogeneity of glomerular changes. Am J Nephrol 1986; 5: 6670 [ Links ]
10. Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin invest 1996; 98: 157588 [ Links ]
11. Massry S, Glassock R. Textbook of Nephrology. 4ta Ed. Baltimore: Williams & Wilkins; 2001.p. 857-865 [ Links ]
12. Feehally J, Johnson R. Comprehensive Clinical Nephrology. 2da Ed. St. Louis: Mosby; 2003.p. 40311 [ Links ]
13. Brenner & Rector's The Kidney. 7ma Ed. Philadelphia: Saunders; 2004.p. 1942 19 [ Links ]
14. Lee HS, Lee JS, Koh HI, Ko KW. Intraglomerular lipid deposition in routine biopsies. Clin. Nephrol 1991; 36: 67-75 [ Links ]
15. Avram MM. Low-density lipoprotein immunofluorescence at the site of renal injury in glomerulosclerosis: a potential pathogenetic role for lipids in renal disease. Am J Kidney Dis 1993; 22: 69-71 [ Links ]
16. Lee HS, Kim YS. Identification of oxidized low density lipoprotein in human renal biopsies. Kidney Int 1998; 54: 848-56 [ Links ]
17. Lee HS, Kruth HS. Accumulation of cholesterol in the lesions of focal segmental glomerulosclerosis. Nephrology 2003; 8: 224-30 [ Links ]
18. Panescu V, Grignon Y, Hestin D, Rostoker G, Frimat L, Renoult E, et al. Recurrence of lecithin cholesterol acyltransferase deficiency after kidney transplantation. Nephrol Dial Transplant 1997, 12: 2430-2. [ Links ]

