










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.32 no.3 Bogotá July/Sept. 2007
Dr. Mauricio Sepúlveda: Residente III Medicina Interna, CES;
Dr. Eduardo Contreras: Medicina Interna, Fundación Valle del Lili, Hospital Universitario del Valle;
Dra. Nataly Martínez: Médica Asistente de Cirugía, Hospital Universitario del Valle. Cali, Valle.
Correspondencia: Dr. Eduardo Contreras. E-mail: edo11@hotmail.com
Recibido: 14/III/07 Aceptado: 30/V/07
Resumen
La enfermedad de Castleman es un desorden raro de mal pronóstico, con compromiso de los ganglios linfáticos del cuerpo. Los sitios más comunes son el tórax, el abdomen y el cuello. Los sitios menos comunes incluyen las axilas, la pelvis y el páncreas. Los crecimientos representan generalmente la ampliación anormal de los ganglios linfáticos encontrados normalmente en estas áreas. Hay dos tipos principales de enfermedad de Castleman: tipo vascular hialino y de células plasmáticas. El tipo vascular hialino explica aproximadamente 90% de las causas. La mayoría de los individuos no exhiben ningún síntoma de esta forma del desorden (asintomático) o pueden desarrollar crecimientos no cancerosos en los nodos de linfa. El tipo de células plasmáticas de enfermedad de Castleman puede asociar a fiebre, pérdida del peso, erupción de piel, destrucción temprana de los eritrocitos, inusualmente anemia hemolítica e hipergammaglobulinemia.
Palabras clave: Castleman, vascular hialino, células plasmáticas.
Abstract
Castleman's disease is a rare disorder with a poor prognosis that may occur in the lymph nodes fine tissues throughout the body. The most common places are the chest, abdomen and the neck. The least common sites include the axilla, pelvis and the pancreas. Usually there is an abnormal enlargement of the lymph nodes found in those sites. There are two main types of Castleman's disease: the hyaline vascular type and the plasma cells type. The hyaline vascular type explains around 90 per cent of the causes. Most of the individuals do not show symptoms of this type of asymptomatic disorder and may develop non cancer type ingrowths in the lymph nodes. The plasma cells type can be associated to fever, weight loss, skin rush, early erythrocytes destruction rarely leading to hemolytic anemia and/or hypergammaglobulinemia.
Key words: Castleman, vascular hyaline, plasma cells.
Caso clínico
Mujer de 43 años de edad, con cuadro clínico de 16 días de evolución consistente en aparición de adenopatías cervicales izquierdas (en zona II) dolorosas a la palpación. Asociado a fiebre entre 38-39ºC y mialgias generalizadas en las cuatro extremidades con incapacidad para la movilización por dolor y pérdida de peso no cuantificada. Recibió tratamiento empírico con antibiótico durante 10 días con poca mejoría de su sintomatología.
Al examen físico de ingreso se encontraba álgica, con limitación funcional por dolor en extremidades, con TA 130/80 mmHg, FC 88x', FR 20x', T 39ºC. Adenopatías unilaterales izquierdas en zona II de cuello, dolorosas a la palpación, fijas, de aproximadamente 1 cm de diámetro, de consistencia dura en forma de conglomerado; amígdalas de tamaño normal, sin lesiones en cavidad oral; cardiopulmonar sin alteraciones; dolor leve a la palpación en abdomen superior, sin hepato ni esplenomegalia; extremidades con dolor intenso a la palpación muscular; sin déficit neurológico aparente.
Se decide iniciar estudio con diagnóstico de síndrome febril persistente y adenopatías. Los paraclínicos mostraron anemia normocítica normocrómica con hemoglobina 11,1 gr/dL, leucocitos 5.600/mm3 (neutrófilos 55% y linfocitos 38%), sin otras alteraciones en el cuadro hemático. Deshidrogenasa láctica 343 U/L. Serología para VIH negativa, VDRL no reactivo, anticuerpos contra citomegalovirus y Eibstein-Barr negativos. El reporte de función hepática, uroanálisis, electrolitos y ácido úrico fue normal con hemocultivos y urocultivo negativos. Electroforesis de proteínas con hipergamaglobulinemia 2.040 mg/dL, ferritina sérica 98 pg/dL. Las imágenes diagnósticas mostraron radiografía de tórax y ecografía abdominal sin alteraciones con TAC cervical que evidenció conglomerado ganglionar (Figura 1).
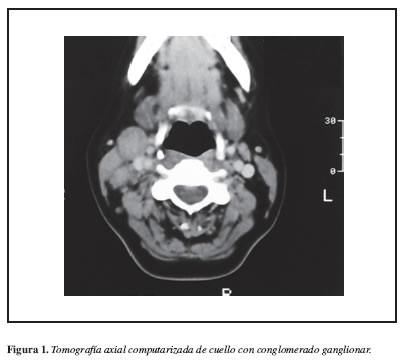
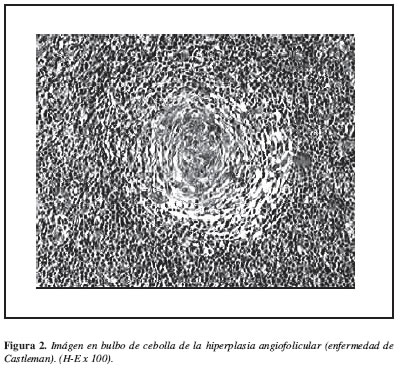
La paciente fue manejada con analgésicos persistiendo con fiebre y mialgias, por lo que se solicitó resección ganglionar obteniendo tejido macroscópicamente sano, un reporte histopatológico compatible con enfermedad de Castleman (Figura 2).
Discusión
La enfermedad de Castleman (o hiperplasia linfonodular angiofolicular) es un desorden linfoproliferativo descrito en 1956 por Benjamín Castleman quien reportó una serie de casos de pacientes con nódulos linfáticos mediastinales hiperplásicos (1). Castleman et al, identificaron dos tipos histológicos; la variante vascular hialina caracterizada por la presencia de pequeños folículos hialinizados no necesariamente con proliferación de los folículos y con marcada proliferación vascular interfolicular. La otra variante descrita de células plasmáticas corresponde a 10-20% de los casos reportados y tiene más centros germinales hiperplásicos y capas de células plasmáticas en las regiones interfoliculares (2). Otro pequeño porcentaje tenía variedad mixta.
La enfermedad de Castleman ha sido asociada con el virus de inmunodeficiencia humana (VIH) y el herpes virus humano 8 (VHH-8), además de otras enfermedades neoplásicas como linfoma Hodgkin, linfoma no Hodgkin, sarcoma de Kaposi y el síndrome de POEMS (polineuropatía, organomegalia, endocrinopatía, gammapatía monoclonal, alteraciones cutáneas) (3).
Tiene clínicamente por lo menos dos comportamientos distintos con tratamiento y pronóstico diferentes. La primera presentación en describirse fue la enfermedad localizada que se denominó enfermedad de Castleman unicéntrica. La presentación multicéntrica fue descrita en 1978 (usualmente con la variante de células plasmáticas) y se manifestó con linfadenopatía periférica generalizada, hepatoesplenomegalia, fiebre y sudoración nocturna (4).
Patología
Se caracteriza por una alteración en la arquitectura ganglionar que permite el reconocimiento de algunas características frecuentes del ganglio afectado. Hay tres variantes principalmente reconocidas:
1. La variante vascular hialina caracterizada por un incremento marcado de folículos anormales con centros germinales atróficos (hialinizados) y zonas amplias periféricas de pequeños linfocitos alrededor de los centros germinales que dan un aspecto de "piel de cebolla". El tejido linfoide interfolicular es hipervascularizado, contiene gran numero de vasos sanguíneos pequeños proliferando. Los sinusoides se encuentran obliterados (1).
2. La variante de células plasmáticas tiene centros germinales hiperplásicos, con folículos hialinos vasculares. La región interfolicular es vascular pero contiene sabanas de células plasmáticas (2).
3. La enfermedad de Castleman con VHH-8 se caracteriza por preservación de la arquitectura nodal y proliferación de folículos que varían de hiperplásicos a involucionados. La región interfolicular contiene vasos sanguíneos proliferantes y células plasmáticas maduras. Se encuentran células plasmáticas variantes – HH-8 e incremento en el número de inmunoblastos en las zonas externas de algunos folículos que pueden invadir los centros germinales. Estas células podrían formar microlinfomas (5).
Enfermedad de Castleman unicéntrica
Es frecuentemente un desorden linfoproliferativo benigno aislado en adultos jóvenes, no se asocia con VHH-8, generalmente curable con resección quirúrgica ganglionar. La edad promedio es de 35 años con igual número de mujeres y hombres. La gran mayoría de pacientes son asintomáticos y la enfermedad fue identificada incidentalemente por estudios imagenológicos. El tamaño en promedio de las lesiones está entre 5 y 7 cm, el 70% se encuentran en mediastino o parahiliares, luego a nivel abdominal y es infrecuente como adenopatías periféricas (2).
Anormalidades en laboratorio se encuentran en menos del 25% de los casos.
De los pacientes con variedad de tipo de células plasmáticas, el 50% presentan anemia, velocidad de sedimentación elevada, hipergammaglobulinemia y plasmocitos en médula ósea.
Tratamiento
La resección completa del nódulo comprometido es curativa, sin que se hayan reportado recurrencias. Los síntomas sistémicos también resuelven (2, 6).
Si la lesión no es completamente resecable, el pronóstico también es favorable con resección parcial, estando asintomáticos por años. La radioterapia puede llevar a remisión parcial o completa con tasas desde el 10 al 40% (6).
La presentación unicéntrica puede estar asociada con incremento en el riesgo de linfomas (linfoma no Hodgkin de celulas B, linfoma Hodgkin) y amiloidosis, a pesar de la resección completa del tumor.
Referencias
1. Belec L, Authier FJ, Mohamed AS, Soubrier M, Gherardi RK. Antibodies to human herpesvirus 8 in POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, skin changes) syndrome with multicentric Castleman's disease. Clin Infect Dis 1999; 28: 678-9. [ Links ]
2. Bowne WB, Lewis JJ, Filippa DA, Niesvizky R, Brooks AD, Burt ME, et al. The management of unicentric and multicentric Castleman's disease: a report of 16 cases and a review of the literature. Cancer 1999; 85:706-17. [ Links ]
3. Castleman B, Iverson L, Menedez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer 1956; 9: 822-30. [ Links ]
4. Dupin N, Diss TL, Kellam P, Tulliez M, Du MQ, Sicard D, et al. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 2000; 95: 1406-12. [ Links ]
5. Iyonaga K, Ichikado K, Muranaka H, Fujii k, Yamaguchi T, Suga M. Multicentric Castleman's disease manifesting in the lung: clinical, radiographic, and pathologic findings and successful treatment with corticosteroid and cyclophosphamide. Intern Med 2003; 42: 182-6. [ Links ]
6. Keller AR, Hochholzer L, Castleman B. Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer 1972; 29: 670-83. [ Links ]
7. Lee FC, Merchant SH. Alleviation of systemic manifestations of multicentric Castleman's disease by thalidomide. Am J Hemato. 2003; 73: 48-53. [ Links ]
8. Peterson BA, Frizzera G. Multicentric Castleman's disease. Semin Oncol 1993; 20: 636-47. [ Links ]
9. Yamasaki S, Tino T, Nakamura M, Henzan H, Ohshima K, Kikuchi M, et al. Detection of human herpesvirus-8 in peripheral blood mononuclear cells from adult Japanese patients with multicentric Castleman's disease. Br J Haematol 2003; 120: 471-7. [ Links ]

