










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.33 no.2 Bogotá Apr./June 2008
(1) Biol. MSc. Profesora Principal. Unidad de Genética. Instituto de Ciencias Básicas. Facultad de Medicina. Universidad del Rosario. Bogotá, Colombia.
(2) MSc. Biol. MSc. Profesora Principal. Unidad de Genética. Instituto de Ciencias Básicas. Facultad de Medicina. Universidad del Rosario. Bogotá, Colombia.
(3) MSc. PhD. Profesor Unidad de Genética. Unidad de Genética. Instituto de Ciencias Básicas. Facultad de Medicina. Universidad del Rosario. Bogotá, Colombia.
Institución donde se realizó el trabajo. Laboratorio de Biología Molecular y Celular. Instituto de Ciencias Básicas. Facultad de Medicina. Universidad del Rosario.
Proyecto Financiado por Colciencias. Cod 1222-04-16333
Correspondencia: Dora Fonseca, Carrera 24 No. 63C-69, Fax: 57-1-3101275. E-mail: dfonseca@urosario.edu.co
Recibido: 29/X/07 Aceptado: 16/IV/08
Resumen
Objetivos: identificación de mujeres portadoras en familias con afectados por distrofia muscular de Duchenne y Becker (DMD/DMB) mediante construcción de haplotipos y determinación de perdida de heterocigocidad.
Introducción: la DMD/DMB es una entidad de herencia recesiva ligada al cromosoma X que se presenta con debilidad muscular, pérdida progresiva de las habilidades motoras y muerte precoz. Son causadas por mutaciones en el gen de la distrofina, el cual contiene 79 exones. Se presentan resultados de dos familias con diagnóstico de DMD/DMB, en las que se había observado previamente deleción en el gen. Se realizó extracción de ADN genómico, seguido de amplificación de 10 STRs, intragénicos y extragénicos del gen de la distrofina, se construyeron haplotipos para los afectados y las mujeres por línea materna en los grupos familiares.
Resultados y conclusiones: con los haplotipos y la determinación de pérdida de heterocigocidad se identificó el estado de portadora en mujeres familiares de afectados con deleción, se discute la aplicación de este método para la identificación de portadoras y su implicación en el asesoramiento genético.
Palabras clave: ADN, deleción, distrofina, distrofia muscular de Duchenne, distrofia muscular de Becker, detección de portadoras.
Abstract
Objectives: identification of carrier women in families affected with Duchenne’s and Becker’s Muscular Dystrophy (DMD/DMB) by means of Haplotypes building and the determination of loss of heterocygocity.
Introduction: DMD/DMB is a recessive inherited disease linked to chromosome X and is presented with muscular weakness, progressive loss of motor skills and early death. These are caused by a mutation of the distrophyne gene that contains 79 axons. These are the results of two families with diagnosis of DMD/DMB, where a deletion of the distrophyne gene there had been previously observed. Extraction of the genomic DNA was performed followed by amplification of intragenic and extragenic10 STRs of the distrophyne gene, haplotypes were built for those affected as well as for the women by maternal line in the family groups.
Results and conclusions: with the haplotypes and the confirmation of loss of heterocygocity we were able to identify the carrier’s status in family women of those affected with deletion. The utilization of this method is being discussed for the identification of carriers and its implications in genetic counseling.
Key words: DNA, deletion, distrophyne, Duchenne’s muscular distrophy, Becker’s muscular distrophy, carrier’s detection.
Introducción
La distrofia muscular de Duchenne (DMD) y su variante menos severa, la distrofia muscular de Becker (DMB), afectan a uno de cada 3.500 varones nacidos vivos en el mundo y se deben a mutaciones en el gen de la distrofina, localizado en el brazo corto del cromosoma X, en Xp21 (1-5). DMD/DMB presentan un modo de herencia ligado al sexo recesivo, por lo que una mujer portadora tendrá un riesgo del 50% de tener hijos afectados o hijas portadoras sanas y, estas últimas, a su vez transmitirán nuevamente la enfermedad a la siguiente generación (6-7).
Publicaciones previas en el mundo muestran que en promedio el 66% de las mutaciones corresponden a deleciones o duplicaciones de exones del gen de la distrofina y el resto corresponde a mutaciones puntuales (8). Las deleciones se distribuyen en dos regiones proclives: la primera, comprendida entre los exones 44 al 52 y la segunda entre los exones 1 al 19 (9). El diagnóstico molecular de los afectados mediante el análisis de 18 exones (de un total de 79 presentes en el gen), permite identificar de manera directa hasta el 98% de las deleciones (10-12). Sin embargo, en Colombia la frecuencia de deleciones es sólo del 31 al 34%(13), aun con el análisis ampliado a 33 exones (datos no publicados). La baja frecuencia de deleciones en nuestro medio, junto con el elevado porcentaje de mutaciones sin identificar, justifican el uso de métodos indirectos, como la construcción de haplotipos y mediante ellos el análisis de la pérdida de la heterocigocidad para identificar mujeres portadoras en las familias con afectados de DMD/DMB.
Un haplotipo es el conjunto de alelos sobre un mismo cromosoma, el cual se puede construir a partir de diversos marcadores genéticos, entre ellos las repeticiones cortas en tandem o STR por su sigla en inglés (Short Tandem Repeats). Estos STR corresponden a una secuencia de 2 a 6 nucleótidos que se repiten uno detrás de otro (ej: ATCATCATCATCATCATC) y se heredan en forma mendeliana simple. Se caracterizan por una gran variabilidad dentro de una misma población y por ello son utilizados en pruebas de identificación humana, medicina forense, microbiología molecular, entre otros.
El concepto de pérdida de heterocigocidad, en la detección de portadoras de deleción de DMD/DMB, hace referencia a la ausencia de uno de los dos alelos que deberían estar presentes en una mujer normal.
En el presente estudio se realizó la construcción de haplotipos y determinación de eventos de pérdida de la heterocigocidad para identificar a las mujeres portadoras de deleción en el gen de la distrofina, familiares de afectados por DMD/DMB. Los haplotipos y la pérdida de heterocigocidad se analizaron mediante STRs extra e intragénicos situados a lo largo del gen.
Material y métodos
Previo diligenciamiento del consentimiento informado, se recolectaron muestras de sangre periférica anticoagulada con ácido etilendiaminotetraacético (EDTA) de 14 individuos pertenecientes a dos familias con afectados de DMD, en quienes previamente se había detectado deleción mediante PCR múltiplex de 17 exones del gen de la distrofina.
Se realizó la extracción del ADN usando el kit comercial GFX®. Se amplificaron, mediante reacción en cadena de la polimerasa (PCR), diez STR: ocho de ellos ubicados dentro del gen de la distrofina y dos (DXS1242 y DXS992) flanqueando los extremos 5’ y 3’ del gen. Se utilizaron controles negativos y positivos de amplificación que garantizaron la calidad del proceso (Tabla 1, Figura 1).
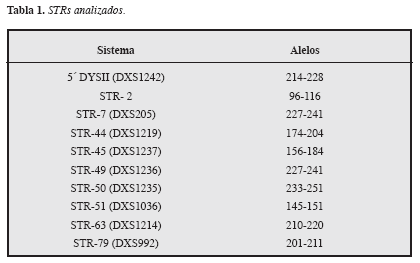
Los amplicones se separaron en geles de secuenciación mediante electroforesis vertical durante 200 minutos a 1500V, 60 mA y 50°C y fueron analizados en un secuenciador ALF-Express. Se asignaron los alelos de acuerdo con el tamaño en pares de bases, según lo publicado en www.dmd.nl/DMD_mPCR.html y se construyeron los haplotipos utilizando la información de los STR analizados para cada persona. Dicha construcción se realiza identificando los alelos presentes en el cromosoma X del afectado lo cual define el “haplotipo ligado a la enfermedad” (haplotipo de riesgo), permitiendo diferenciar el cromosoma X que contiene el gen de la distrofina mutado. Posteriormente, se construyeron los haplotipos en los demás miembros de la familia y aquellas mujeres en las que se identificó el haplotipo de riesgo se consideraron como posibles portadoras.
La pérdida de heterocigocidad se identifica por la ausencia de amplificación del(os) STR(s) situados en el segmento delecionado y representan un método directo para identificar mujeres portadoras. El afectado no mostrará amplificación de estos STR.
Resultados
Familia DMD7
El caso índice presenta deleción de los exones 12 al 45 (13). El análisis de 10 STR permitió la construcción del haplotipo en el afectado y los demás miembros de la familia. El haplotipo del cromosoma X presente en el afectado fue denominado XA y mostró una ausencia de amplificación para los STR I-44 (DXS1219) e I-45( DXS1237), ambos incluidos en el segmento delecionado (Figura 2). La madre presentó haplotipos para cada cromosoma X denominados XA y XB; si se tiene en cuenta que DMD/DMB presenta una herencia ligada al X recesiva, se esperaría que la madre del afectado fuera portadora de la deleción observada en el hijo que fue la causante del desarrollo de la enfermedad, es decir, el XA materno ligado con la enfermedad debería indicar pérdida de heterocigocidad y por lo tanto ausencia de los alelos para los STRs I44 (DXS1219) e I-45 (DXS1237) en dicho cromosoma. Este resultado esperado no se observó y en el cromosoma XA materno estaban presentes los alelos 198 y 180 para los STR I44 e I45 respectivamente (Figuras 2 y 4), lo que indica que la madre no tiene deleción de este segmento del gen de la distrofina y por tanto la deleción en el afectado es producto de una mutación “de novo”.
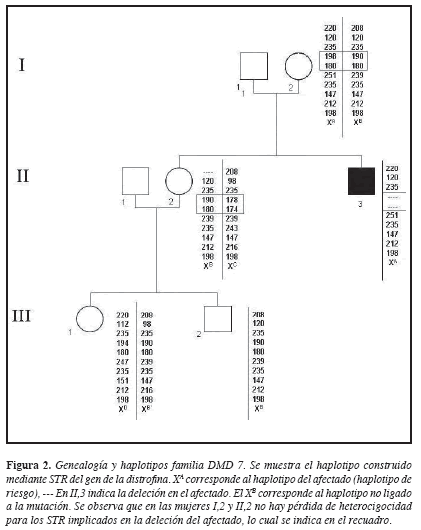
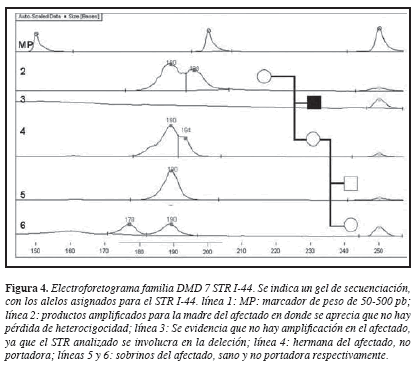
La hermana II,2, presentó haplotipos llamados XB y XC, que indican su estado de no portadora, ya que heredó el haplotipo de no riesgo de su madre (XB) y el XC sano de su padre.
Familia DMD108
El caso índice II,5; presentó deleción de los exones 50-52. El análisis de 10 STR mostró en el haplotipo llamado XA, ausencia de amplificación para los STR I-49 (DXS1236), I-50 (DXS1235) e I-51(DXS1036), que están incluidos en el segmento delecionado (Figura 3). La madre del paciente I,1 y una de las hermanas II,1, presentaron un haplotipo idéntico al del afectado (XA), la ausencia de amplificación para uno de los dos alelos en estos STR permiten determinar la pérdida de heterocigocidad, confirmando su estado de portadora de la enfermedad (Figuras 3 y 5).
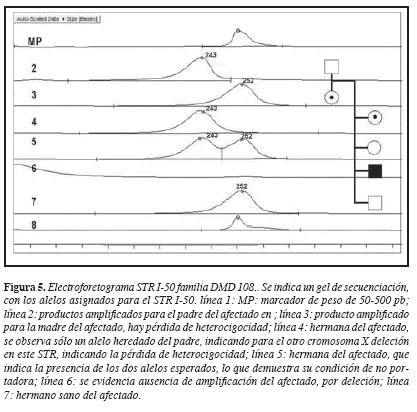
Las hermanas II,3 y II,4, heredaron el haplotipo materno no ligado a la enfermedad (XB), descartándolas como portadoras (Figura 3).
Discusión
Las DMD/DMB son entidades frecuentes, progresivas y letales, para las que no se ha logrado, hasta la fecha, un tratamiento efectivo. Por tener un modo de herencia ligado al X recesivo, las mujeres portadoras tendrán un riesgo del 50% de hijos varones afectados e hijas portadoras. La prevención primaria de DMD/DMB se basa en identificar e informar a las mujeres el riesgo de heredar la enfermedad a su descendencia.
La determinación del estado de portadora de deleciones se puede abordar por diferentes estrategias que tratan de obviar el problema metodológico que genera la presencia de una copia normal del gen de la distrofina, en las portadoras. Por esta razón algunos autores han utilizado la técnica Hibridación Fluorescente In situ (FISH), mediante sondas específicas que señalan la presencia o ausencia de uno o varios exones sobre el cromosoma X, sin embargo, ésta sólo es útil cuando el paciente afectado presenta una deleción confirmada (14). Estudios moleculares previos desarrollados por nuestro grupo de investigación han indicado que sólo el 31% de los pacientes presentan deleciones (13), por lo que el FISH es una alternativa poco informativa para la población colombiana de afectados y sus familiares. Otro método de identificación de portadoras consiste en el análisis de la dosis génica, a través de densitometría o mediante PCR en tiempo real (15), esta última es la más sensible y específica, pero en la actualidad su acceso es limitado.
El análisis del gen de la distrofina mediante STR, es una alternativa que permite la detección de portadoras de DMD/DMB de manera indirecta o directa. En el presente estudio se analizaron 10 STR que permitieron construir haplotipos e identificar los cromosomas X de los afectados y sus familias. La detección de portadoras mediante análisis indirecto se realiza identificando el cromosoma X ligado a la enfermedad, a través de la construcción de haplotipos, permitiendo reconocer y rastrear en las familiares del afectado por línea materna, si portan o no el cromosoma con el haplotipo de riesgo, indicando con un 80% de certeza el estado de portadora o no portadora (sensibilidad dada por los posibles eventos de recombinación del gen). En la detección directa mediante STR, la construcción del haplotipo analizado junto con el hallazgo de la pérdida de heterocigocidad, logra identificar con un 100% de certeza las mujeres portadoras de deleción del gen.
En la familia DMD7, el afectado (II,3), mostró en su único cromosoma X el haplotipo XA, en donde no se observó amplificación para los STRs I-44 e I-45, localizados en los intrones 44 y 45, ambos incluidos en la región afectada por la deleción (exones 12 al 45), observada en estudios previos (13). A la madre del afectado (I,2) quien no tenía historia familiar de DMD/DMB, se le pudo identificar como no portadora de la deleción en células sanguíneas, porque, a pesar de compartir con el afectado el haplotipo XA, ella mostró amplificación de los STR I-44 e I-45. Sin embargo, este hallazgo no descarta completamente el estado de portadora, por posibles eventos de mosaicismo gonadal, los cuales consisten en la presencia de una mutación de novo en el gen de la distrofina, que está sólo en algunos de los gametos, mientras que otros portarán un gen de la distrofina normal. Los eventos de mosaicismo gonadal se han informado en la literatura con una frecuencia hasta del 20% (16). La identificación de estos eventos debe tenerse en cuenta en el asesoramiento genético, por cuanto el riesgo para las mujeres con mosaicismos no es predecible, ya que no se conoce el porcentaje de óvulos portadores de la mutación. La hermana del afectado (II,2), no presentó ni el haplotipo ligado a la mutación, ni deleciones, por lo que se le identificó como una mujer no portadora, al igual que la sobrina del afectado, III,1 (Figura 4).
En la familia DMD108, el caso índice II,5, presentó deleción de los exones 50 al 52 y el haplotipo ligado a la mutación se denominó XA. La madre (I,1), presentó el haplotipo XA con la ausencia de amplificación de los STR I-49, I-50 e I-51, confirmándose como portadora de la mutación causal de DMD. El análisis del árbol genealógico completo indica que el cromosoma XA que porta la deleción fue heredado por el abuelo materno del afectado, es decir, la mutación en la familia tuvo su origen en sus espermatozoides. Esta observación es acorde con lo descrito en la literatura que indica que los espermatozoides sufren frecuentemente mutaciones de novo, que determinan estados de mosaicismo gonadal que generan enfermedades genéticas de herencia recesiva ligada al X (17). En el análisis de la hermandad del afectado se identificó a una hermana (II,1) como portadora de la mutación al poseer el haplotipo XA y dos hermanas más no portadoras II,3 y II,4.
De esta manera se demuestra que la construcción de haplotipos junto con el análisis de pérdida o no de heterocigocidad son útiles para establecer el estado o no de portadora en familias con DMD/DMB. Otros estudios han mostrado hallazgos similares, detectando portadoras y mutaciones de novo, a través de la construcción de haplotipos intra y extragénicos (18, 19).
Agradecimientos
Los autores agradecen a los pacientes y sus familias y a la Asociación Colombiana de Distrofia Muscular (ACDM). El presente trabajo fue financiado por Colciencias, bajo el código 122-04-16333.
Referencias
1. Duchenne GB. De l’ataxie locomotrice progressive. Archives générales de médecine. Paris ; 1858, 5 sér., 12: 641-652; 13: 36-62, 158-1881, 417-451. [ Links ]
2. Gowers W. A Manual of disease of the Nervous System. J Ment Sci 1887; 32: 594-6. [ Links ]
3. Darras BT. Molecular genetics of Duchenne and Becker muscular Dystrophy. J Pediatr 1990; 117: 1-15. [ Links ]
4. Emery AE. Oxford monographs on medical genetics. Duchenne Muscular Dystrophy. Oxford: Oxford University Press; 1993: p.539-63. [ Links ]
5. Nevo Y, Muntoni F, Sewry C, Legum C, Kutai M, Harel S, et al. Large in-frame deletions of the rod-shaped domain of the dystrophin gene resulting in severe phenotype. Isr Med Assoc J 2003; 5: 94-7. [ Links ]
6. Emery AE, Skinner R, Holloway S. A study of possible heterogeneity in Duchenne muscular dystrophy. Clin Genet 1979; 1: 444-9. [ Links ]
7. Erazo Torricelli R. Actualización en distrofias musculares. Rev Neurol 2004; 39: 860-71. [ Links ]
8. Den Dunnen JT, Grootscholten PM, Bakker E, Blonden LAJ, Ginjaar HB, Wapenaar MC, et al. Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and CDNA analysis of 194 cases reveals 116 deletions and 13 duplications. Am J Hum Genet 1989; 45: 835-47. [ Links ]
9. Shomrat R, Gluck E, Legun C, Shiloh Y. Relatively low proportion of Dystrophin Gene deletions in Israeli Duchenne and Becker Muscular Dystrophy patients. Am J Med Genet 1994; 49: 369-73. [ Links ]
10. Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 1988; 16: 11141–56. [ Links ]
11. Beggs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98-percent DMD/DMB gene deletions by polymerase chain reaction. Hum Genet 1990; 86: 45-8. [ Links ]
12. Montejo-Pujadas Y, Zaldivar-Vaillant T, Acevedo-Lopez AM. Diagnostic techniques described in the study of Duchenne/Becker muscular dystrophy. Rev Neurol 2002; 33: 278-81. [ Links ]
13. Silva CT, Fonseca D, Restrepo CM, Contreras N, Mateus H. Deleciones en el gen de la Distrofina en 62 familias colombianas: correlación genotipo-fenotipo para la distrofina muscular de Duchenne y Becker. Colombia Médica (en línea) 2004 Oct-Dic [consultado 2004 octubre 10]; 35:191-198. Disponible en: http://colombiamedica.univalle.edu.co/Vol35No4/BODY/contenido.htm [ Links ]
14. Ligon AH, Kashork CD, Richards CS, Shaffter LG. Identification of female carriers for Duchenne and Becker muscular dystrophies using a FISH-based approach. Eur J Hum Genet 2000; 8: 293-8. [ Links ]
15. Joncourt F, Neuhaud B, Jostarndt K, Kleinle S, Steiner B, Gallati S. Rapid Identification of female carriers of DMD/BMD by Quantitative Real Time PCR. Hum Mutat 2004; 23: 385-91. [ Links ]
16. Ferreiro V, Szijan I, Giliberto F. Detection of germline mosaicism in two Duchenne muscular dystrophy families using polymorphic dinucleotide (CA)n repeat loci within the dystrophin gene. Mol Diagn 2004; 8: 115-21. [ Links ]
17. Delgado-Luengo W, Borjas-Fuentes L, Zabala-Fernandez W, Fernández-Salgado E, Solís-Añez E, Chavez C, et al. Detección de portadoras de Distrofia Muscular de Duchenne/Becker a través del análisis de loci STRs ligados al gen de la distrofína en familias venezolanas. Invest Clin 2002; 43: 239-54. [ Links ]
18. Mukherjee M, Chaturvedi L, Srivastava S, Mittal RD, Mittal B. De novo mutations in sporadic deletional Duchenne muscular dystrophy (DMD) cases. Exp Mol Med 2003; 35: 113-17. [ Links ]
19. Kim UK, Cho MS, Chae JJ, Kim SH, Hong SS, Lee SH, et al. Allelic Frequencies of six (CA)n microsatellite markers of the Dystrophin gene in the Korean population. Hum Hered 1999; 49: 205-7. [ Links ]

