










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkActa Medica Colombiana
versão impressa ISSN 0120-2448
Acta Med Colomb v.34 n.2 Bogotá abr./jun. 2009
(1)TM. Profesor Asociado, Director Programa de Investigación en Factores de Riesgo de Enfermedades Cardiovasculares (PIFRECV). Facultad Ciencias de la Salud, Universidad de Talca, Chile;
(2) TM. Dr, Profesor Asistente, Depto. Bioquímica Clínica e inmunohematología, Facultad Ciencias de la Salud, Universidad de Talca, Chile;
(3) TM. Dr(c). Profesor Asistente, Depto. Bioquímica Clínica e inmunohematología, Facultad Ciencias de la Salud, Universidad de Talca, Chile;
(4) PhD, Profesor Asociado, Escuela de Medicina, Facultad de Ciencias de la Salud, Universidad Católica del Maule, Chile;
(5) Médico. Programa de Diabetes, Servicio de Medicina, Hospital Regional de Talca y Profesor, Depto. Bioquímica Clínica e inmunohematología, Facultad Ciencias de la Salud, Universidad de Talca, Chile;
(6) Médico. Unidad de Cirugía Vascular, Servicio de Cirugía, Hospital Regional de Talca, Chile.
Correspondencia. Dr. Iván Palomo G., TM, Departamento de Bioquímica Clínica e Inmunohematología, Facultad Ciencias de la Salud, Universidad de Talca, Talca, Chile. Casilla: 747, Talca, Chile. Teléfono-Fax: 56-71-200488 E-mail: ipalomo@utalca.cl
Recibido 05/XII/08 Aceptado 06/V/09
Resumen
El síndrome metabólico (SM) se caracteriza por la presencia de tres de los siguientes criterios: obesidad abdominal, hipertrigliceridemia, disminución del colesterol HDL, presión arterial alta en tratamiento, e hiperglicemia o diabetes mellitus en tratamiento. En la mayoría de los países desarrollados y en vías de desarrollo, la prevalencia de SM fluctúa entre 20 y 30% en la población adulta.
La presencia de SM representa un aumento del riesgo cardiovascular, situación que se asociaría en parte, al estado protrombótico que acompaña este síndrome, y que incluye disfunción endotelial, hipercoagulabilidad, hipofibrinólisis y activación plaquetaria.
El estado protrombótico del SM no está suficientemente estudiado. Seguramente el mayor conocimiento de los mecanismos fisiopatológicos que contribuyen a dicho estado, podría ser importante en la búsqueda de nuevas estrategias de prevención y tratamiento de eventos cardiovasculares en dichos pacientes.
Palabras claves: síndrome metabólico, estado protrombótico, disfunción endotelial.
Abstract
Metabolic syndrome (MS) is characterized by the presence of three of the following criteria: abdominal obesity, hypertriglyceridemia, decreased HDL cholesterol levels, high blood pressure under treatment, and hyperglycemia or diabetes mellitus under treatment. In most developed and developing countries, the prevalence of MS fluctuates between 20% and 30% in adult population.
The presence of MS represents an increased cardiovascular risk. This situation appears to be associated in part with the prothrombotic state that accompanies this syndrome, which includes endothelial dysfunction, hypercoagulability, hypofibrinolysis and platelet activation.
The prothrombotic state of MS has not been sufficiently studied. Better knowledge of the pathophysiological mechanisms that contribute to such state is surely to be important in the search for new preventive and therapeutic strategies for cardiovascular events in such patients.
Key words: metabolic syndrome, prothrombotic state, endothelial dysfunction.
Introducción
El síndrome metabólico (SM) es una constelación de factores de riesgo cardiovascular de origen metabólico (1), caracterizado según el Adult Treatment Panel III (ATP III), por la presencia de tres de los siguientes criterios: obesidad abdominal (circunferencia de cintura: hombres >102 cm, mujeres >88 cm), hipertrigliceridemia (>150 mg/dl), disminución del HDL colesterol (HDL-c) (hombres <40; mujeres <50 mg/dl), hipertensión arterial (>130/85 mm Hg) o en tratamiento con fármacos antihipertensivos, e hiperglicemia (=100 mg/dl) o en tratamiento de diabetes mellitus (DM) (2-4). Está establecido que el SM representa un mayor riesgo cardiovascular (5-7).
La obesidad visceral y la insulinorresistencia (con hiperinsulinemia compensatoria) son la base del SM; factores que favorecen o aumentan esta condición son el sedentarismo, la edad avanzada, y factores genéticos y endocrinos. El SM es una condición progresiva que puede incluir desde niveles límite hasta alteraciones categóricas en los factores de riesgo (5, 8).
La prevalencia del SM varía según edad, género, origen étnico y estilo de vida (9, 10), y los criterios diagnósticos utilizados ATPIII o International Diabetes Federation (IDF) (11). En la población adulta la prevalencia de SM fluctúa entre 20 y 30% (12, 13). En Chile, la Encuesta Nacional de Salud (ENS-2003) mostró una prevalencia de 22,6% sin diferencias entre hombres y mujeres, pero muy dependiente de la edad: 17-24 años (4.6%), 25-44 años (17.9%), 45-64 años (36.5%) y sobre 65 años (48%) (14). Nuestro grupo encontró que en adultos de la ciudad de Talca, la prevalencia de SM fue de 29.5% y 36.4%, según criterios ATPIII e IDF respectivamente (15). Por otra parte, previamente determinamos en la misma población, que la frecuencia de factores de riesgo cardiovascular clásicos fue levemente superior a la encontrada en la ENS-2003 (16).
La obsesidad se asocia con aumento de ácidos grasos libres circulantes, lo que induce aumento de la excreción hepática de glucosa y consecuentemente produce hiperglicemia, la que a su vez induce hiperinsulinemia como respuesta compensatoria. Este proceso es mediado y regulado por señales y respuestas desde el receptor de la insulina, lo que se deteriora cada vez más en la medida que se perpetúa el trastorno metabólico, situación que se conoce como insulinorresistencia (17, 18).
Desde el punto de vista fisiopatológico, el SM además de desarrollar hiperglicemia, dislipidemia e hipertensión arterial, se caracteriza por presentar un estado protrombótico que en parte, explicaría el mayor riesgo cardiovascular que presentan los individuos con SM (19, 20).
Estado protrombótico del SM
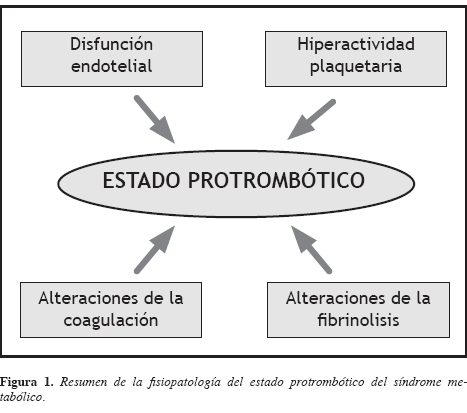
Los antecedentes actuales indican que el estado protrombótico incluye alteraciones del endotelio, de la coagulación y de la fibrinolisis, y activación plaquetaria (19, 20) (Figura 1).
Disfunción endotelial
Actualmente el endotelio no sólo es reconocido como una barrera física entre la sangre y la pared vascular, sino como un importante órgano con múltiples funciones endocrinas y paracrinas (21). En condiciones fisiológicas, el endotelio a través de varias moléculas bioactivas, regula la contracción vascular, la adhesión de leucocitos, el crecimiento de células musculares lisas y la agregación plaquetaria (22, 23).
Se entiende por disfunción endotelial a una serie de alteraciones que afectan la síntesis, liberación, difusión o degradación de los factores que se generan en el endotelio (24). Entre las alteraciones asociadas a la disfunción endotelial en el SM se encuentran: expresión de moléculas de adhesión (25), disminución de síntesis de óxido nítrico (NO) (26) y de prostaciclina (PGI2) (27), aumento de liberación de endoperóxidos (28, 29), aumento de producción de especies reactivas del oxígeno (ROS) (28, 29), aumento de secreción de endotelina 1 (ET-1) (30) y disminución de la sensibilidad del músculo liso vascular a los vasodilatadores de origen endotelial (28). Dicho de otra manera, disfunción endotelial corresponde a una pérdida parcial o completa del balance entre factores vasoconstrictores (ET-1 y angiotensina II) y vasodilatadores (NO y PGI2), factores promotores e inhibidores del crecimiento, factores proaterogénicos y antiaterogénicos, factores protrombóticos (PAI-1) y antitrombóticos (PGI2 y heparán sulfato) (23).
Varios mecanismos asociados a la insulinorresistencia han sido implicados en el desarrollo de disfunción endotelial (30). Entre otros aspectos, se han incluido los fenómenos inflamatorios y la producción de ROS (31). Algunas adipoquinas favorecen la disfunción endotelial: TNF-a, aumentado en el SM (32-34), inhibe la acción de la lipoproteinlipasa, activa el estrés oxidativo y aumenta la síntesis de proteínas de fase aguda (35); e IL-6 activa las células endoteliales (CE) (36) y aumenta la expresión de factor tisular (FT) en monocitos (37). Entre las moléculas de adhesión celular, VCAM-1 (Vascular cell adhesion molecule-1), puede ser regulada in vitro en respuesta a TNF-a (38); la concentración sérica de la forma soluble de VCAM-1 (sVCAM-1) se encuentra aumentada en sujetos con SM (39) y en pacientes hipertensos no compensados (40). La expresión de ICAM-1 (Intercelullar adhesion molecule-1) en CE es inducible por IL-1, TNF-alfa e IFN-y; y E-Selectina por IL-1ß, TNF-alfa (41). Nosotros hemos confirmado el aumento de los niveles séricos de sVCAM-1 en individuos con SM (42). Los ácidos grasos libres circulantes, aumentados en el SM, también participan en el deterioro del endotelio (43).
Los individuos con SM presentan niveles circulantes de micropartículas de origen plaquetario, eritrocitario, leucocitario y endotelial, superiores a los encontrados en personas sin SM (44). En los individuos con SM, dicha situación se asocia con una disminución del NO y aumento del estrés oxidativo, y como consecuencia favorece el establecimiento de disfunción endotelial (44).
Alteraciones de la coagulación
En lo que se refiere a la coagulación, en el SM se han encontrado niveles elevados de fibrinógeno (45), lo que también fue observado por nosotros (46). Además se han encontrado niveles aumentados de tres de los factores vitamina K dependientes (FVII, FIX y FX) (47), factor XIII (47) y factor von Willebrand (47). La hiperfibrinogenemia se explica principalmente porque las adipoquinas IL-6 y TNFa inducen la síntesis de proteínas de fase aguda como fibrinógeno y proteína C reactiva (PCR) a nivel hepático (48).
Alteraciones de la fibrinolisis
En cuanto al sistema fibrinolítico, el SM se caracteriza principalmente por aumento del inhibidor del activador del plasminógeno 1 (PAI-1) (46, 49), un reactante de fase aguda sintetizado por los adipocitos y las plaquetas (50), y cuya concentración plasmática se relaciona con la cantidad de grasa visceral (51) y con el polimorfismo de su región promotora -675 4G/5G (52). El plasma de sujetos con SM forma coágulos más densos que los individuos sin SM (53, 54). Por otra parte, las personas con SM presentan tiempos de lisis del coágulo más prolongados que los individuos sin SM (53), lo que se explicaría principalmente por los altos niveles de PAI-1 (55).
Hiperactividad plaquetaria
Las plaquetas en reposo, de forma discoide y 1,5-3 µm de diámetro, normalmente no se unen al endotelio normal, pero sí al endotelio dañado (56). Al ser activadas, junto con cambiar de forma, desarrollan varios procesos, entre otros: cambio conformacional de la GPIIb-IIIa que une fibrinógeno y FVW, expresión de P-selectina y factor tisular, liberación de agonistas plaquetarios (ADP, TxA2 y serotonina), PF4 y factores de la coagulación, entre otras moléculas. En resumen, ocurren los fenómenos de adhesión, secreción y agregación plaquetaria, todo lo cual también favorece la coagulación y unión de leucocitos (56).
La contribución de las plaquetas al estado protrombótico del SM se ha relacionado a una respuesta aumentada de las mismas (57-62), observándose algunos marcadores de activación plaquetaria y liberación de micropartículas plaquetarias (63), todos hallazgos relacionados al proceso de formación del trombo arterial (64). Se han descrito algunas alteraciones en las plaquetas de individuos con SM, las que podrían favorecer su activación: disminución de la fluidez de su membrana, posiblemente producto de cambios en su composición lipídica; aumento del metabolismo del ácido araquidónico con incremento en la producción de TxA2 y aumento de calcio libre intracelular (61). La presencia de LDLox también podría contribuir a la activación plaquetaria, ya que éstas presentan receptores de LDLox (CD36 y LOX-1) (65, 66). Otro hallazgo descrito en las plaquetas de individuos con SM es que presentan aumento de su volumen, lo que podría servir como marcador de activación plaquetaria (67).
Se ha descrito que la hipertrigliceridemia y los ácidos grasos libres favorecen la agregación plaquetaria in vitro (68). Por otra parte, la hiperactividad plaquetaria que se observa en pacientes con SM se ha relacionado directamente con el nivel sérico de adiponectina (69-72) y de leptina (73-75); para esta última existen receptores en las plaquetas (76, 77).
En el SM las plaquetas podrían tener participación precoz en el proceso aterogénico en asociación con la disfunción endotelial. Se ha descrito que las plaquetas tienen algún rol en el proceso inflamatorio de la aterogénesis (78-80). Así, inicialmente la unión de plaquetas al endotelio activado (disfuncional) ocurriría a través de las moléculas P-selectina/ PSGL-1 (P-selectin glycoprotein ligand-1), luego por unión GPIb/FVW y posteriormente vía integrinas (GPIIb-IIIa/ ICAM-1 o aVβ3). Así las plaquetas se activan y secretan moléculas que favorecen el proceso inflamatorio propio de la aterogénesis, favoreciendo la activación del endotelio, y reclutando monocitos y otras plaquetas. Entre dichas moléculas se pueden mencionar:
- IL-1 y CD40L participan en la alteración de las CE, las que luego liberan MCP-1 (monocyte chemoatractant protein); los monocitos reclutados expresan ICAM-1, molécula que favorece la unión de leucocitos, todo lo cual favorece el fenómeno inflamatorio. Hemos encontrado que sCD40L se encuentra aumentado en individuos con SM (42).
- PF4 activa a los linfocitos T vía quimioquina CXCR3; así estimula la secreción de citoquinas y favorece la inflamación (81).
- RANTES (CCL5) actúa como quimioatractante para monocitos (82, 83).
- PDGF (Platelet-derived growth factor) estimula la proliferación de células musculares provocando hiperplasia de la capa íntima de la pared vascular y también tiene una función quimiotáctica para monocitos, actuando como amplificador de la respuesta inflamatoria (84).
- ENA-78 (Epithelial neutrophil activating peptide-78, CXCL5) participa en el reclutamiento de monocitos al sitio de la lesión (85).
- ADP, TxA2 y serotonina, entre otras, favorecen el reclutamiento de más plaquetas (56), lo que podría amplificar el fenómeno.
Los antecedentes anteriores indican que las plaquetas, en un contexto de disfunción endotelial, pueden contribuir al proceso inflamatorio de la aterogénesis (79, 80, 86-88). Siendo la aterogénesis del SM un proceso inflamatorio que se desarrolla a largo plazo, es probable que las plaquetas participen.
Comentario final
Es probable que el estado protrombótico presente en individuos con SM sea más complejo que lo descrito hasta ahora. La participación precoz de las plaquetas en la aterogénesis, en el contexto de la disfunción endotelial, podría ser importante. Se requieren estudios que caractericen el estado protrombótico del SM en humanos y en modelos murinos, abordando las alteraciones endoteliales, en la coagulación, la fibrinólisis y las plaquetas. El conocimiento que se obtenga podría ayudar en la búsqueda de nuevas estrategias de prevención y tratamiento de eventos cardiovasculares en sujetos con SM.
Referencias
1. Barrera M, Pinilla, A, Cortés E, Mora G, Rodríguez M. Síndrome Metabólico: una mirada interdisciplinaria. Rev Colomb Cardiol 2008; 15: 111-26. [ Links ]
2. Nathan DM, Davidson MB, DeFronzo RA, Heine RJ, Henry RR, Pratley R, et al. Impaired fasting glucose and impaired glucose tolerance: implications for care. Diabetes Care 2007; 30: 753-9. [ Links ]
3. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001; 285: 2486-97. [ Links ]
4. Mendivil C. Obesidad y Síndrome Metabólico. Acta Med Colomb 2005; 30: 164-7. [ Links ]
5. Grundy SM. Metabolic syndrome: connecting and reconciling cardiovascular and diabetes worlds. J Am Coll Cardiol 2006; 47: 1093-100. [ Links ]
6. Merchán A. Síndrome Metabólico y riesgo de enfermedad cardiovascular. Acta Med Colomb 2005; 30: 150-4. [ Links ]
7. Saito I, Iso H, Kokubo Y, Inoue M, Tsugane S. Metabolic syndrome and allcause and cardiovascular disease mortality. Circ J 2009; 878-84. [ Links ]
8. Manzur f, Alvear C, Alayón A. Caracterización fenotípica y metabólica del síndrome metabólico en Cartagena de Indias. Rev Colomb Cardiol 2008; 15: 97-101. [ Links ]
9. Batsis JA, Nieto-Martinez RE, Lopez-Jimenez F. Metabolic syndrome: from global epidemiology to individualized medicine. Clin Pharmacol Ther 2007; 82: 509-24. [ Links ]
10. Lombo B, Satizábal C, Villalobos C, Tique C, Kattah W. Prevalencia del Síndrome Metabólico en pacientes diabéticos. Acta Med Colomb 2007; 32: 9-15. [ Links ]
11. Alberti KG, Zimmet P, Shaw J; IDF Epidemiology Task Force Consensus Group. The metabolic syndrome--a new worldwide definition. Lancet 2005; 366: 1059-62. [ Links ]
12. Grundy SM. Metabolic syndrome pandemic. Arterioscler Thromb Vasc Biol 2008; 28: 629-36. [ Links ]
13. Manzur F, de la Ossa M, Trespalacios E, Abuabara Y, Lujan M. Prevalencia del Síndrome metabólico en el municipio de Arjona, Colombia. Rev Colomb Cardiol 2008; 15: 215-22. [ Links ]
14. Ministerio de Salud de Chile. Encuesta Nacional de Salud 2003. Departamento de Salud Pública de la Pontificia Universidad Católica de Chile. Informe Técnico. 2003. [ Links ]
15. Mujica V, Leiva E, Icaza G, Diaz N, Arredondo M, Moore-Carrasco R, et al. Evaluation of metabolic syndrome in adults of Talca city, Chile. Nutr J 2008; 7: 14. [ Links ]
16. Palomo GI, Icaza NG, Mujica EV, Núñez FL, Leiva ME, Vásquez RM, et al. Prevalence of cardiovascular risk factors in adult from Talca, Chile. Rev Med Chil 2007; 135: 904-12. [ Links ]
17. Boden G, She P, Mozzoli M, Cheung P, Gumireddy K, Reddy P, et al. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-kappaB pathway in rat liver. Diabetes 2005; 54: 3458-65. [ Links ]
18. Griffin ME, Marcucci MJ, Cline GW, Bell K, Barucci Lee D, et al. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999; 48: 1270-4. [ Links ]
19. Palomo I, Alarcón M, Moore-Carrasco R, Argilés JM. Hemostasis alterations in metabolic syndrome (review). Int J Mol Med 2006; 18: 969-74. [ Links ]
20. Alessi MC, Juhan-Vague I. Metabolic syndrome, haemostasis and thrombosis. Thromb Haemost 2008; 99: 995-1000. [ Links ]
21. Baumgartner-Parzer SM, Waldhäusl WK. The endothelium as a metabolic and endocrine organ: its relation with insulin resistance. Exp Clin Endocrinol Diabetes 2001; 109: 166-79. [ Links ]
22. Chen K, Pittman RN, Popel AS. Nitric oxide in the vasculature: where does it come from and where does it go? A quantitative perspective. Antioxid Redox Signal 2008; 10: 1185-98. [ Links ]
23. Esper RJ, Nordaby RA, Vilariño JO, Paragano A, Cacharrón JL, Machado RA. Endothelial dysfunction: a comprehensive appraisal. Cardiovasc Diabetol 2006; 5: 4. [ Links ]
24. Giannotti G, Landmesser U. Endothelial dysfunction as an early sign of atherosclerosis. Herz 2007; 32: 568-72. [ Links ]
25. Schram MT, Stehouwer CD. Endothelial dysfunction, cellular adhesion molecules and the metabolic syndrome. Horm Metab Res 2005; 37: 49-55. [ Links ]
26. Bulló M, Casas-Agustench P, Amigó-Correig P, Aranceta J, Salas-Salvadó J. Inflammation, obesity and comorbidities: the role of diet. Public Health Nutr 2007; 10: 1164-72. [ Links ]
27. Xiang L, Naik JS, Hodnett BL, Hester RL. Altered arachidonic acid metabolism impairs functional vasodilation in metabolic syndrome. Am J Physiol Regul Integr Comp Physiol 2006; 290: 134-8. [ Links ]
28. Meerarani P, Badimon JJ, Zias E, Fuster V, Moreno PR. Metabolic syndrome and diabetic atherothrombosis: implications in vascular complications. Curr Mol Med 2006; 6: 501-14. [ Links ]
29. Roberts CK, Sindhu KK. Oxidative stress and metabolic syndrome. Life Sci 2009; 84: 705-12. [ Links ]
30. Wheatcroft SB, Williams IL, Shah AM, Kearney MT. Pathophysiological implications of insulin resistance on vascular endothelial function. Diabet Med 2003; 20: 255-68. [ Links ]
31. King GL, Loeken MR. Hyperglycemia-induced oxidative stress in diabetic complications. Histochem Cell Biol 2004; 122: 333-8. [ Links ]
32. Ferroni P, Basili S, Falco A, Davì G. Inflammation, insulin resistance, and obesity. Curr Atheroscler Rep 2004; 6: 424-31. [ Links ]
33. López-Jaramillo P, Pradilla LP, Bracho, Y. Papel del adipocito en la inflamación del síndrome metabólico. Acta Med Colomb 2005; 30: 85-91. [ Links ]
34. Gurrola-Díaz CM, Sánchez-Enriquez S, Oregon-Romero E, García-López PM, Garzón de la Mora P, Bastidas-Ramírez BE, et al. Establishment of a cut-point value of serum TNF-alpha levels in the metabolic syndrome. J Clin Lab Anal 2009; 23: 51-6. [ Links ]
35. Chu NF, Spiegelman D, Hotamisligil GS, Rifai N, Stampfer M, Rimm EB. Plasma insulin, leptin, and soluble TNF receptors levels in relation to obesityrelated atherogenic and thrombogenic cardiovascular disease risk factors among men. Atherosclerosis 2001; 157: 495-503. [ Links ]
36. Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW. C-reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol 1999; 19: 972-8. [ Links ]
37. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW, Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112: 1796-808. [ Links ]
38. Troseid M, Lappegard KT, Mollnes TE, Arnesen H, Seljeflot I. Changes in serum levels of E-selectin correlate to improved glycaemic control and reduced obesity in subjects with the metabolic syndrome. Scand J Clin Lab Invest 2005; 65: 283-90. [ Links ]
39. Kougias P, Chai H, Lin PH, Yao Q, Lumsden AB, Chen C. Effects of adipocytederived cytokines on endothelial functions: implication of vascular disease. J Surg Res 2005; 126: 121-9. [ Links ]
40. Palomo I, Marín P, Alarcón M, Gubelin G, Viñambre X, Mora E, et al. Patients with essential hypertension present higher levels of sE-selectin and sVCAM-1 than normotensive volunteers. Clin Exp Hypertens 2003; 25: 517-23. [ Links ]
41. Dinarello CA. The IL-1 family and inflammatory diseases. Clin Exp Rheumatol 2002; 20: 1-13. [ Links ]
42. Palomo I, Jaramillo J, Alarcón M, Gutiérrez C, Moore-Carrasco R, Segovia F, et al. Soluble vascular cell adhesion molecule-1 and soluble CD40L increased concentration appears in individuals with metabolic syndrome. Molecular Medicine Reports 2009; 2: 481-5. [ Links ]
43. Wang XL, Zhang L, Youker K, Zhang MX, Wang J, LeMaire SA, et al. Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes 2006; 55: 2301-10. [ Links ]
44. Agouni A, Lagrue-Lak-Hal AH, Ducluzeau PH, Mostefai HA, Draunet- Busson C, Leftheriotis G, et al. Endothelial dysfunction caused by circulating microparticles from patients with metabolic syndrome. Am J Pathol 2008; 173: 1210-9. [ Links ]
45. Mertens I, Van Gaal LF. Visceral fat as a determinant of fibrinolysis and hemostasis. Semin Vasc Med 2005; 5: 48-55. [ Links ]
46. Palomo I, Gutiérrez C, Alarcón M, Jaramillo J, Segovia F, Leiva E, et al. Increased Concentration of Plasminogen Activator Inhibitor-1 and fibrinogen in Individuals with metabolic syndrome. Molecular Medicine Reports. 2009; 2: 253-7. [ Links ]
47. Devaraj S, Rosenson RS, Jialal I. Metabolic syndrome: an appraisal of the proinflammatory and procoagulant status. Endocrinol Metab Clin North Am 2004; 33: 431-53. [ Links ]
48. Isezuo SA. The metabolic syndrome: Review of current concepts. Niger Postgrad Med J 2006; 13: 247-55. [ Links ]
49. Skurk T, Hauner H. Obesity and impaired fibrinolysis: role of adipose production of plasminogen activator inhibitor-1. Int J Obes Relat Metab Disord 2004; 28: 1357-64. [ Links ]
50. Aso Y. Plasminogen activator inhibitor (PAI)-1 in vascular inflammation and thrombosis. Front Biosci 2007; 12: 2957-66. [ Links ]
51. Aso Y, Wakabayashi S, Yamamoto R, Matsutomo R, Takebayashi K, Inukai T. Metabolic syndrome accompanied by hypercholesterolemia is strongly associated with proinflammatory state and impairment of fibrinolysis in patients with type 2 diabetes: synergistic effects of plasminogen activator inhibitor-1 and thrombinactivatable fibrinolysis inhibitor. Diabetes Care 2005; 28: 2211-6. [ Links ]
52. Kinik ST, Ozbek N, Yuce M, Yazici AC, Verdi H, Atac FB. PAI-1 gene 4G/5G polymorphism, cytokine levels and their relations with metabolic parameters in obese children. Thromb Haemost 2008; 99: 352-6. [ Links ]
53. Carter AM, Cymbalista CM, Spector TD, Grant PJ; EuroCLOT Investigators. Heritability of clot formation, morphology, and lysis: the EuroCLOT study. Arterioscler Thromb Vasc Biol 2007; 27: 2783-9. [ Links ]
54. Collet JP, Allali Y, Lesty C, Tanguy ML, Silvain J, Ankri A, et al. Altered fibrin architecture is associated with hypofibrinolysis and premature coronary atherothrombosis. Arterioscler Thromb Vasc Biol 2006; 26: 2567-73. [ Links ]
55. Alessi MC, Juhan-Vague I. PAI-1 and the metabolic syndrome: links, causes, and consequences. Arterioscler Thromb Vasc Biol 2006; 26: 2200-7. [ Links ]
56. Santos MT, Aranda E, Vallés J, Palomo I. Hemostasia Primaria. En: Palomo I, Pereira J, Palma J, eds. Hematología: Fisiopatología y Diagnóstico. Talca: Editorial Universidad de Talca; 2005.cap.19. [ Links ]
57. Trovati M, Anfossi G. Insulin, insulin resistance and platelet function: similarities with insulin effects on cultured vascular smooth muscle cells. Diabetologia 1998; 41: 609-22. [ Links ]
58. Anfossi G, Trovati M. Pathophysiology of platelet resistance to anti-aggregating agents in insulin resistance and type 2 diabetes: implications for anti-aggregating therapy. Cardiovasc Hematol Agents Med Chem 2006; 4: 111-28. [ Links ]
59. Davi G, Guagnano MT, Ciabattoni G, Basili S, Falco A, Marinopiccoli M, et al. Platelet activation in obese women: role of inflammation and oxidant stress. JAMA 2002; 288: 2008-14. [ Links ]
60. Trovati M, Anfossi G. Influence of insulin and of insulin resistance on platelet and vascular smooth muscle cell function. J Diabetes Complications 2002; 16: 35-40. [ Links ]
61. Arteaga RB, Chirinos JA, Soriano AO, Jy W, Horstman L, Jimenez JJ, et al. Endothelial microparticles and platelet and leukocyte activation in patients with the metabolic syndrome. Am J Cardiol 2006; 98: 70-4. [ Links ]
62. Basili S, Pacini G, Guagnano MT, Manigrasso MR, Santilli F, Pettinella, et al. Insulin resistance as a determinant of platelet activation in obese women. J Am Coll Cardiol 2006; 48: 2531-8. [ Links ]
63. Andres E, Goichot B. Metabolic syndrome and venous thrombosis: potential role of microparticles. Haematologica 2007; 92: 64-5. [ Links ]
64. Darvall KA, Sam RC, Silverman SH, Bradbury AW, Adam DJ. Obesity and thrombosis. Eur J Vasc Endovasc Surg 2007; 33: 223-33. [ Links ]
65. Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med 2007; 13: 1086-95. [ Links ]
66. Valiyaveettil M, Kar N, Ashraf MZ, Byzova TV, Febbraio M, Podrez EA. Oxidized high-density lipoprotein inhibits platelet activation and aggregation via scavenger receptor BI. Blood 2008; 111: 1962-71. [ Links ]
67. Tavil Y, Sen N, Yazici HU, Hizal F, Abaci A, Cengel A. Mean platelet volume in patients with metabolic syndrome and its relationship with coronary artery disease. Thromb Res 2007; 120: 245-50. [ Links ]
68. Englyst NA, Taube JM, Aitman TJ, Baglin TP, Byrne CD. A novel role for CD36 in VLDL-enhanced platelet activation. Diabetes 2003; 52: 1248-55. [ Links ]
69. Gualillo O, González-Juanatey JR, Lago F. The emerging role of adipokines as mediators of cardiovascular function: physiologic and clinical perspectives.Trends Cardiovasc Med 2007; 17: 275-83. [ Links ]
70. Kato H, Kashiwagi H, Shiraga M, Tadokoro S, Kamae T, Ujiie H, et al. Adiponectin acts as an endogenous antithrombotic factor. Arterioscler Thromb Vasc Biol 2006; 26: 224-30. [ Links ]
71. Elbatarny HS, Netherton SJ, Ovens JD, Ferguson AV, Maurice DH. Adiponectin, ghrelin, and leptin differentially influence human platelet and human vascular endothelial cell functions: implication in obesity-associated cardiovascular diseases. Eur J Pharmacol 2007; 558: 7-13. [ Links ]
72. von Hundelshausen P, Weber C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ Res 2007; 100: 27-40. [ Links ]
73. Beltowski J. Leptin and atherosclerosis. Atherosclerosis 2006; 189: 47-60. [ Links ]
74. Konstantinides S, Schäfer K, Koschnick S, Loskutoff DJ. Leptin-dependent platelet aggregation and arterial thrombosis suggests a mechanism for atherothrombotic disease in obesity. J Clin Invest 2001; 108: 1533-40. [ Links ]
75. Konstantinides S, Schafer K, Loskutoff DJ. The prothrombotic effects of leptin possible implications for the risk of cardiovascular disease in obesity. Ann N Y Acad Sci 2001; 947: 134-41. [ Links ]
76. Elbatarny HS, Maurice DH. Leptin-mediated activation of human platelets: involvement of a leptin receptor and phosphodiesterase 3A-containing cellular signaling complex. Am J Physiol Endocrinol Metab 2005; 289: 695-702. [ Links ]
77. Giandomenico G, Dellas C, Czekay RP, Koschnick S, Loskutoff DJ. The leptin receptor system of human platelets. J Thromb Haemost 2005; 3: 1042-9. [ Links ]
78. Siegel-Axel DI, Gawaz M. Platelets and endothelial cells. Semin Thromb Hemost 2007; 33: 128-35. [ Links ]
79. Lindemann S, Krämer B, Daub K, Stellos K, Gawaz M. Molecular pathways used by platelets to initiate and accelerate atherogenesis. Curr Opin Lipidol 2007; 18: 566-73. [ Links ]
80. Palomo I, Toro C, Alarcón M. Rol of the platelets in the pathophysiology of atherosclerosis. Molecular Medicine Reports 2008; 1: 179-84. [ Links ]
81. Mueller A, Meiser A, McDonagh EM, Fox JM, Petit SJ, Xanthou G, et al. CXCL4-induced migration of activated T lymphocytes is mediated by the chemokine receptor CXCR3. J Leukoc Biol 2008; 83: 875-82. [ Links ]
82. Schober A, Zernecke A. Chemokines in vascular remodeling. Thromb Haemost 2007; 97: 730-7. [ Links ]
83. Schober A, Manka D, von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, et al. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation 2002; 106: 1523-9. [ Links ]
84. Tang J, Kozaki K, Farr AG, Martin PJ, Lindahl P, Betsholtz C, et al. The absence of platelet-derived growth factor-B in circulating cells promotes immune and inflammatory responses in atherosclerosis-prone ApoE-/- mice. Am J Pathol 2005; 167: 901-12. [ Links ]
85. Baltus T, von Hundelshausen P, Mause SF, Buhre W, Rossaint R, Weber C. Differential and additive effects of platelet-derived chemokines on monocyte arrest on inflamed endothelium under flow conditions. J Leukoc Biol 2005; 78: 435-41. [ Links ]
86. Sakkinen PA, Wahl P, Cushman M, Lewis MR, Tracy RP. Clustering of procoagulation, inflammation, and fibrinolysis variables with metabolic factors in insulin resistance syndrome. Am J Epidemiol 2000; 152: 897-907. [ Links ]
87. Lindemann S, Krämer B, Seizer P, Gawaz M. Platelets, inflammation and atherosclerosis. J Thromb Haemost 2007; 5: 203-11. [ Links ]
88. May AE, Langer H, Seizer P, Bigalke B, Lindemann S, Gawaz M. Plateletleukocyte interactions in inflammation and atherothrombosis. Semin Thromb Hemost 2007; 33: 123-7. [ Links ]

