










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActa Medica Colombiana
versión impresa ISSN 0120-2448
Acta Med Colomb v.34 n.3 Bogotá jul./sep. 2009
Una perspectiva histórica desde la semiología dermatológica hasta la correlación genética
A historical perspective, from dermatologic semiology to genetic correlation
(1) Servicio de Medicina Interna, Hospital Occidente de Kennedy, Clínica Universitaria Carlos Lleras Restrepo, Universidad Nacional de Colombia, Bogotá D.C.; (2)Servicio de Nefrología y Diálisis, Hospital El Tunal E.S.E; Servicio de Terapia Renal, Hospital Universitario Clínica San Rafael, Bogotá D.C., Colombia.
Correspondencia: Dr. Juan Pablo Camargo, Cra. 79 G No 35-81 Sur, Teléfono fijo: (571) 4520952, Teléfono móvil: (57) 311-4676538, Bogotá D.C., Colombia
E-mail: jpcamargome@unal.edu.co - battement2002@yahoo.com
Recibido: 26/VI/09 Aceptado: 22/VII/09
Resumen
Objetivos: rescatar los aspectos fundamentales en el desarrollo histórico de la enfermedad de Fabry. Identificar las áreas de la medicina que contribuyeron al conocimiento actual de la enfermedad. Contribuir en la formación médica en el contexto de la construcción del conocimiento a través de la historia.
Material y métodos: revisión de la literatura desde 1898 a 2009 a través de Pubmed, Embase, Ovid, Science Direct, SpringerLink, Blackwell Synergy, MD Consult, ProQuest, Highwire, biblioteca y hemeroteca de la Universidad Nacional de Colombia.
Resultados: se encontraron 2181 artículos en relación con la enfermedad de Fabry, los cuales fueron seleccionados en idiomas inglés y alemán con énfasis en revisiones históricas médicas, informe de casos o serie de casos relevantes, estudios clínicos, revisiones sistemáticas y revisiones en general. Del total de artículos, seleccionamos aquellos que tuvieran mayor interés desde el punto de vista histórico y de aplicación clínica, por lo que se consideró finalmente para nuestro análisis, 66 fuentes bibliográficas.
Conclusiones: la enfermedad de Fabry aunque rara y poco conocida, muestra el curso de la observación clínica a la configuración fisiopatológica y genética, en un contexto multiorgánico siendo un reto para el clínico actual. Aunque sus descripciones iniciales son del norte europeo, se puede evidenciar el gran aporte de la literatura americana en su entendimiento molecular y terapéutico, y en las dos últimas décadas los japoneses, con nuevas formas de presentación y variedades cromosómicas.
Palabras clave: Fabry, alfa-galactosidasa A, globotriaosilceramida, terapia de reemplazo enzimático.
Abstract
Objectives: 1) To highlight the core issues in the historical development of Fabry's disease. 2) To identify the areas of medicine that have contributed to current knowledge of the disease. 3) To participate in medical education in the context of the construction of knowledge.
Materials and methods: a review of the literature from 1898 to 2009 through Pubmed, Embase, Ovid, Science Direct, SpringerLink, Blackwell Synergy, MD Consult, ProQuest, HighWire, and the library and archive of the National University of Colombia.
Results: 2.181 articles concerning Fabry's disease were found. Articles in English and German were selected, laying emphasis on historical revisions, case reports or series of important cases, clinical trials, systematic reviews, and reviews in general. Of all the items selected, 66 were taken into account for our analysis, due to their historical interest and clinical application.
Conclusions: Fabry disease, although rare and little known, shows the course from clinical observation to genetic and pathophysiological settings, in a multi-context that continues to be a challenge for clinicians today. While initial descriptions are from Northern Europe, we can show the great contribution of American literature to the understanding of the molecular and therapeutic aspects of the disease. During the last two decades, Japanese investigators have identified new clinical presentations and chromosomal variations.
Key words: Fabry, alpha-galactosidase A, globotriaosylceramide, enzyme replacement.
Introducción
La enfermedad de Fabry también conocida como angioqueratoma corporis diffusum es un trastorno hereditario debido a mutaciones en el gen de la a-galactosidasa (a-Gal), situado en el cromosoma X (Xq22.I), produciendo un déficit de su actividad enzimática. Esto condiciona el depósito de glicoesfingolípidos neutros (principalmente globotriaosilceramida-Gb) en los lisosomas de las células endoteliales, periteliales, del músculo liso y entre otras, con su consecuente elevación en plasma. Se calcula una incidencia aproximada de 1:40.000 a 60.000 en hombres, no obstante, algunas poblaciones refieren tasas que van desde 1:80.000 nacidos vivos a 1:116.000. La edad promedio de muerte es de 41 años con informes de supervivencia a los 60 años (1, 2).
El fenotipo clásico es observado con mayor frecuencia en hombres y raramente en mujeres. Manifestaciones tempranas de la enfermedad como acroparestesias, angiokeratoma e hipohidrosis llegan a ser aparentes en la edad pediátrica. Complicaciones a nivel renal, cardiaco y en sistema nervioso central aparecen en etapas posteriores (3).
Se describe acerca de 300 mutaciones que generan alteración en el plegamiento de la enzima, sus sitios de unión o del seguimiento del proceso de maduración de la misma. Para que ocurra debe haber una alteración importante de la actividad enzimática ya que con una función entre 5% y 10%, se previenen las manifestaciones clínicas (4). En esta revisión se analizarán los principales hechos y contribuciones que han permitido reconocer y tratar esta interesante enfermedad.
Principales gestores en el nacimiento y reconocimiento de la enfermedad
William Anderson
Nació en 1842 en Londres, fue educado en la Escuela de Londres. En un principió estudió arte, llegando a ser pupilo de la famosa Escuela de Lambeth de las escuelas de arte donde fue condecorado. No obstante, consideró estudiar medicina, ingresando al St. Thomas Hospital en 1864, donde fue entrenado por los cirujanos John Simon Le y Gros Clark y los anatomistas Jones y Rainey. Ganó el primer premio universitario de la sociedad de Cheseldan, siendo promovido en el colegio médico. Regresó a St. Thomas en 1871 como registrador quirúrgico y como asistente en el grupo de anatomía aprovechando sus habilidades en el dibujo (Figura 1).

De 1873 a 1880 se traslada a Japón como director médico de la Escuela Médica Naval de Tokio. Luego regresa a St. Thomas para continuar en el grupo de anatomía y como asistente de cirugía. Posterior a esto, se encargó del nuevo Departamento de Piel y de los servicios del paciente ambulatorio. En 1891 fue nombrado cirujano haciendo parte del honorable grupo quirúrgico y trabajando la mayor parte de su vida, salvo el tiempo en que hizo parte en la Universidad de Londres. Su última posición fue la de profesor de la Academia Real de Anatomía. De igual forma, continuó desarrollando actividades artísticas, configurando el perfil de un hombre muy talentoso y sensible. Falleció en el año 1900 (5).
Johannes Fabry
Nació en 1860 en Jülich cerca de Aix la Chapelle. Fue educado en el Jülich y Düren. También considerado un hombre sensible y talentoso por ser exponente del violín y la viola. Decidió estudiar medicina y posteriormente cirugía en las universidades de Bonn y Berlín, culminando sus estudios en 1884. Un accidente en que perdió su pierna, desgraciadamente lo privó de continuar su entrenamiento como cirujano. Como consecuencia, decidió estudiar dermatología, una nueva disciplina médica en ese momento, en el Dermatological Prusiano Real de la Universidad de Bonn donde Doutrelepont se hizo su maestro. En 1886 escribió su primera revisión y a finales de su vida en 1930 había escrito más de 70. En 1889 fue el director designado del Departamento de Dermatología de Dortmund, siendo en 1919 profesor honorario, terminando su compromiso como presidente en 1928 y muriendo allí por una septicemia (5) (Figura 2).

Robert J Desnick
Profesor y Jefe del Departamento de Genética y Ciencias Genómicas en Mount Sinai School of Medicine y jefe médico del Departamento de Genética Médica y Genómica en el Hospital Mount Sinai. Recibió su BA, Ph.D, MD y grados de la Universidad de Minnesota, inmediatamente fue nombrado profesor asociado de pediatría, biología celular y genética. En 1977, se incorporó a la facultad de Mount Sinai School of Medicine con Arthur J. Cohen y Nellie Z. Profesor de pediatría y genética, y jefe de la división de genética médica y molecular. En 1993, se convirtió en el primer jefe del Departamento de Genética Humana (recientemente rebautizado Genética y Ciencias Genómicas) en Mount Sinai School of Medicine.
Es miembro de numerosas sociedades científicas, incluyendo la Sociedad Americana de Pediatría, American Society for Clinical Investigation y la Asociación Americana de Médicos. Fue director de la Junta Americana de Genética Médica (ABMG), diplomático de la Fundación Colegio Americano de Genética Médica (ACMG) y miembro de la Junta de Directores de la Fundación (ACMG). Ha colaborado en los NIH (Consejo Consultivo Nacional para el Centro Nacional de Recursos para Investigación). Es fundador y ex presidente de la asociación de profesores de derechos humanos y de genética médica. Es ex presidente del Consejo Académico de Sociedades de la Asociación Americana de Colegios Médicos (AAMC) y actualmente es presidente de la AAMC (Figura 3).

Ha realizando numerosos aportes en el entendimiento y manejo de enfermedades como la porfiria y la enfermedad de Fabry. Esta última con 100 artículos en la literatura indexados en Pubmed para esta revisión y considerándose autoridad en este campo.
Año 1898 primera descripción de Fabry
Anderson publica la primera descripción del angiokeratoma corporis diffusum en The British Journal of Dermatology (Figura 4). Se trataba de un paciente de 39 años de ocupación pintor, quien a los 11 años de edad presentó un eritema en la cara anterior de rodillas, posteriormente las lesiones se extendieron al tronco, extremidades superiores y el resto de los miembros inferiores, con una extensión máxima a los 17 años (Figura 5). Presentó un episodio de sangrado intestinal a los 18 años. De igual manera desarrolló un neuroma en cadera derecha y venas varicosas en las extremidades.

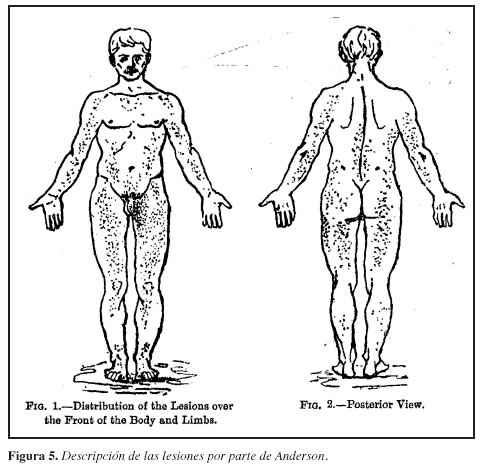
Anderson describió angiectasias multicapilares generalizadas, como tumores pequeños o prominencias subalterno-epidérmicas, algunos con formas verrucosas en el techo de la epidermis. Las angiectasias las describía como el tamaño de la cabeza de un alfiler, hasta de semilla de cáñamo, con una estructura papulosa cubriendo la superficie de la piel salvo la cara, palmas y plantas de los pies, con mayor presencia en el escroto.
Los cambios de nonpapulosos más pequeños eran similares a las mordeduras de la pulga (pero no presentaban comezón); los más grandes eran en racimos, sin configuración específica. Por otra parte, Anderson mencionaba la asociación de sus hallazgos con la presencia de proteinuria intermitente (6).
En el mes de abril de este mismo año, Fabry describe su primer paciente (Figura 6). Se trataba de un adolescente de 14 años, quien cuatro años antes había desarrollado erupciones cutáneas en la rodilla izquierda, las cuales se extendían a extremidades superiores y el tronco. Posteriormente el paciente desarrolló dilatación capilar en su rodilla derecha. De forma simultánea el paciente presentaba pérdida de peso y disminución de la capacidad física. Al examen, Fabry encontró mucosas pálidas, párpados y labios edematosos, signos de consolidación en el hemitórax superior izquierdo, múltiples nódulos dolorosos en cuello y región inguinal, la piel evidenció erupciones agrupadas del tamaño de una cabeza de alfiler y otras más grandes eran papulosas de color azul y negro y pequeños capilares dilatados en comisuras de la boca. En el examen sanguíneo y de orina, no informó anormalidades. Dos meses después el paciente presentó un episodio de hemoptisis y epistaxis. Dos meses después de estos episodios el paciente permaneció asintomático. Se realizó biopsia de piel que mostró hipertrofia de estrato córneo, elongación papilar, aumento y agrupamiento de capilares rodeados de linfocitos y células plasmáticas con extravasación de eritrocitos. En el interrogatorio, Fabry informó que el abuelo paterno de este paciente falleció de una patología renal no clara (7).

El doctor Fabry fue recopilando casos e información previa, que permitieron análisis y entendimiento de la enfermedad que tomaría su nombre, ejemplo de esto es una revisión de su autoría. En ésta, explica la evolución y distinción de los angiokeratomas nombrados en la época: "akroasphycticum y corporis naeviforme" y resume lo que los autores previos describían con los hallazgos de sus investigaciones, no sólo mencionando una descripción clínica, sino también etiológica (8) (Figura 7).

Año 1909
Steiner and Voerner describen los síntomas neurológicos asociados a la enfermedad (9).
Año 1925
Weicksel describe los cambios a nivel cardiaco, oftálmico y algunos aspectos hereditarios de la enfermedad. Aunque son en la mayoría correlaciones, son bastante aceptados para la época (10).
Año 1939
Ruiter and Pompen describen algunos de sus pacientes, cuyas características clínicas coincidían con algunas informes de la literatura (11).
Año 1947
En el estudio anatomopatológico de uno de sus pacientes, Ruiter y cols identificaron los cambios en el sistema vascular asociado de una sustancia ligada a un péptido (12).
Año 1950
Un patólogo de Hamburgo ratificó lo encontrado por Ruitery cols. Posterior a una autopsia encontró en la histopatología a través de luz polarizada, estructuras que denominó como birefringent Maltese- cross-like, como una característica típica. Por otra parte, Kuehnau, un bioquímico de Hamburgo, caracterizó estas estructuras como glicofosfolípidos (13).
Año 1951
Hornbostel publica la información en cuanto a la descripción de síntomas de 17 pacientes informados en el mundo. El 100% presentaba cambios en piel, 71% cambios urinarios, 47% calambres, 41% compromiso cardiaco, 35% hipertensión arterial y 29 % opacidades corneales (14).
Año 1955
Por parte del Departamento de Medicina de la Universidad de Utah, informan el caso de un paciente cuya clínica era confusa y encajaba con muchos síndromes, especialmente con púrpura y telangiectasia hereditaria. Después de indagar en la literatura se consideró que era una forma de angiokeratoma que cumplía las características con lo descrito inicialmente en 1898. Es el primer informe americano en la literatura (15).
Año 1958
Dos patólogos de la Universidad de Wiirzburg describen los hallazgos patológicos y bioquímicos que mostrarían que la enfermedad es sistémica con alteración de toda la musculatura lisa del corazón y de los nervios autónomos. Caracterizan el almacenamiento de lípidos y grupos celulares como en el riñón y el sistema reticuloendotelial. Recalcan el papel de la trihexoceramida y cómo el defecto de la enzima desempeña un papel importante en el metabolismo de estas células (16) (Figura 8).

Por otra parte dos hechos importantes pero contradictorios sucedieron en este año. Por su parte, Ruiter describió que la enfermedad de Fabry sólo afectaba a hombres, mientras Colley informó la posibilidad de compromiso en el sexo femenino (17,18).
Año 1959
Karr recopila la información de 30 pacientes, todos de sexo masculino y en los cuales se demostró aumento de esfingomielinas como fenómeno esencial. Karr concluyó que la enfermedad era una perturbación hereditaria del metabolismo de los lípidos (19).
Año 1962
Hoffman y Hauser mencionan las manifestaciones neurológicas de la enfermedad. Se discute la posibilidad de trombosis cerebral, vértigo, hemiparesia, diplopía, disartria y hemiataxia. De igual forma realizan la aproximación a la hipoacusia neurosensorial (20).
Año 1963
Sweeley aisló dos nuevos tipos de glicolípidos: glucosilfingosina y galactosilfingosina (21).
Año 1964
Groot recopiló la información de 45 pacientes, incluyendo siete de su casuística (cuatro hombres y tres mujeres que tenían sólo síntomas ligeros), y que pertenecían a una familia de cuatro generaciones. También se refirió a las estructuras nubladas de los huesos, necrosis del intestino y diarrea. Continuó caracterizando el corporis del angiokeratoma diffusum como una perturbación hereditaria dominante del metabolismo de lípidos. Se refirió al compromiso en el sexo femenino, cuya presentación está determinada por tipificación genética que se aclarará en futuras investigaciones (22).
Año 1965
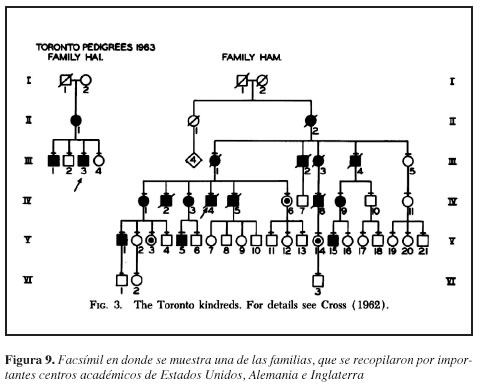
Con la asistencia técnica de Colette Wifflerl, se realiza una recopilación de casos de diferentes hospitales universitarios entre ellos Wisconsin, Royal Free Hospital, Instituto Lister, Clínica Mayo, Toronto East General Hospital y Dermatologische Universiteitskliniek en Amsterdam. Con la información recolectada se realizan pruebas de herencia para el cromosoma X. Los datos sobre la vinculación entre el locus Xg y el locus angiokeratoma corporis diffusum sugieren que estos dos genes pueden ser mensurables en
distancia el uno del otro con la mejor estimación de la fracción de recombinación (23) (Figura 9).
Hashimoto a través de la microscopía electrónica, observó la presencia de cuerpos en el endotelio y células en el músculo liso, fibrocitos y células perivasculares, las cuales caracterizó como lisosomas. Los cuerpos endoteliales reaccionaban enzimáticamente con ácido de fosfatasa y se refirió a ellos como "lisosomas apiñados". Concluyó que había un disturbio en la actividad enzimática lisosomal por una anormalidad genética (24). Por otra parte, Dempsey después de investigar una familia con cuatro afectados con Fabry y otros cuatro con alta probabilidad de estarlo, concluyó que la enfermedad estaba ligada al sexo por gen deficiente de forma ocasional en mujeres heterocigóticas y penetrancia constante en hombres homocigotos. De igual forma, informó la distribución de los lípidos en los eritrocitos, niveles elevados de colesterol, aumento en la concentración de fosfolípidos, cefalinas y disminución de los niveles de lecitina y esfingomielina (25).
En este mismo año, Opitz informó que el defecto era localizado en el brazo largo del cromosoma X. Concluyó que los hombres eran los principalmente afectados y que éstos tenían un defecto genético exclusivo del sexo femenino y las abuelas no influían en sus nietos. Identificó 11 hijas cuyos padres eran portadores de la enfermedad de Fabry (23).
Año 1967
Roscoe Brady y cols demostraron la deficiencia de la actividad de ceramidatrihexosidasa en la mucosa intestinal de cada dos hombres homocigotos con enfermedad de Fabry. Consideraron que la baja actividad no era atribuible a la presencia de un inhibidor de la actividad enzimática y tampoco a la presencia de cofactores. Se ratifica que la ceramidatrihexosidasa se libera como componente intermedio de la degradación de globosidasa. En este trabajo se corrobora que el defecto extremo del clivaje de la enzima en homocigotos explica la inhabilidad de los pacientes para depósito. Por otra parte, las observaciones de los heterocigotos, donde las manifestaciones clínicas eran discretas, sugieren menor depósito de glucolípidos y por tanto el complemento enzimático es suficiente. Este trabajo fue la base para el desarrollo de la terapia de reemplazo enzimático (26).
Por parte del Departamento de Patología de la Universidad de California informan el caso inusual de un hombre de 27 años con historia familiar no clara que a pesar de una amplia evidencia de enfermedad generalizada dada por lesiones en la piel, anormalidades oculares y proteinuria, la función renal estaba mínimamente afectada. Estudios de microscopía electrónica del riñón y piel mostró que la mayoría de las células tenían las características de los lípidos descritas en la enfermedad de Fabry (27).
Año 1969
Philippart informó la distribución de los glicolípidos en la orina de los pacientes con Fabry. Encontró que todos los hombres tenían incremento de ceramida-hexosidas y que trihexosidas se elevaban más que dihexosidas. Sin embargo, en las mujeres las ceramida-hexosidas se encontraban disminuidos, con trihexosidas disminuidas al igual que las dihexosidas (28).
Año 1971
Rady y Desnick propusieron la posibilidad y necesidad de diagnóstico prenatal de la enfermedad (29, 30).
Año 1972
Desnick y cols describieron la resolución de la enfermedad después del trasplante renal (31). Por otra parte, Pilz y cols describen el caso de un joven de 21 años con alteraciones neuromusculares tanto en la clínica como en la electromiografía. Exámenes posteriores sugirieron miopatía debido al depósito de glucolípidos en el músculo esquelético y que la neuropatía era debida a disturbios vasculares. Con este caso se realiza una revisión sobre las manifestaciones neurológicas de la enfermedad de Fabry siendo base para las descripciones actuales (32) (Figura 10).

Año 1973
Brady describe el control de la enfermedad después de la terapia con ceramidatrihexosidasa. Desde aquí se determina el concepto de terapia de reemplazo enzimático, para insuficiencia y no para deficiencia enzimática completa (33).
En este mismo año, Víctor Mckusick publica una revisión sobre el trabajo del doctor Edward Alfred Cockayne (Figuras 11 y 12), un médico que se dedicó a las enfermedades en niños, particularmente las hereditarias, aportando el papel de los factores genéticos en la piel, describiendo tres categorías de la determinación genética: cromosomal, monogénica y poligénica. Aportó razonamientos en cuanto a las variedades de angiokeratomas con posibles hipótesis para su presentación que hoy en día son bases para preguntas no resueltas (34).
Año 1974
El grupo de patología del Instituto der Friedrich-Schiller, informan en detalle tres autopsias de miembros de una familia. Uno de ellos del sexo femenino. La típica lipidosis era demostrada por histoquímica y por microscopía electrónica tanto por técnica ultrafina como por congelación. Los depósitos de sustancias tenían características ultraestructurales compatibles con trihexosidoceramida y dihexosidaceramida, los cuales son típicos de la enfermedad de Fabry (35).
Año 1976
Pierides investigó a una familia de tres hombres que sufrieron angiokeratoma corporis diffusum. Uno de ellos presentó distintivamente pocos signos de la enfermedad. Análisis cromosómico mostró un genotipo XYY. Duplicación del cromosoma y proporciona en este caso la producción suficiente de la enzima y previene al paciente de la enfermedad severa. Recíprocamente, individuos que sufren de síndrome de Down tienen la constelación cromosómica XXY y nunca son portadores de enfermedad de Fabry (36).
Año 1978
Johnson y Desnick investigaron los mecanismos fisiopatológicos del depósito en Fabry a través de cultivos de venas de homocigotos con la enfermedad, comparados con venas umbilicales de recién nacidos sin la enfermedad. Encontraron que los primeros tenían numerosas inclusiones citoplasmáticas, probablemente repletas de sustratos de lisosomas. Por otra parte, encontraron propiedades similares de la a-GAL A informada en trabajos previos. Concluyeron que los homocigotos con Fabry que acumulaban trihexosidoceramida podrían alterar el aparato lisosomal a través de receptores mediados por lipoproteínas y se acumulaban por la actividad defectuosa de la a-GAL A (37).
Año 1979
Se presentan las manifestaciones oculares en una serie de 37 homocigotos hombres y 25 mujeres heterocigotas con la enfermedad de Fabry. Los hallazgos oculares no afectaron la visión, pero se consideraron únicos y de diagnóstico. Depósitos corneales se observaron en casi todos los pacientes con mayor gravedad que en los heterocigotos. El cristalino tomaba una coloración crema, con depósitos capsulares a nivel anterior, a veces con distribución en forma de "hélice" en una tercera parte de los homocigotos y en ninguno de los heterocigotos. Una tenue pero única opacidad capsular posterior con un patrón de ramificacines radiales se observó en 37% de la homocigotos y 14% de los heterocigotos. Dilataciones conjuntivales en forma de buque y tortuosidad retiniana fueron más frecuentes y graves en los homocigotos. Pérdida visual grave se produjo en dos homocigotos como resultado de oclusión unilateral de arterias centrales (38).
Año 1980
Se informa un caso de un hombre cuyos niveles plasmáticos de glicofosfolípidos se determinaron por cromatografía líquida de alta resolución y al cual se le realizaron tres sesiones de plasmaféresis día, disminuyendo los niveles de trihexosidoceramida a rangos normales. Aproximadamente 70 mumoles de la enzima fueron removidos en dos sesiones. Este caso fue base para muchos trabajos como una forma de inicio del reemplazo enzimático (39). De igual forma se informan siete pacientes con la enfermedad, los cuales fueron evaluados por la presencia y el grado de obstrucción de las vías respiratorias. Se encontró que todos tenían obstrucción importante del flujo aéreo. Además, la evaluación de las células epiteliales obtenidas por broncoscopia demostraron que estas células con cuerpos de inclusión figuraban en consonancia con los depósitos de trihexosidoceramida, lo que sugiere que parte de su flujo de aire a la obstrucción funcional puede ser secundaria a enfermedad de las vías respiratorias intrínsecas. Aunque todos los sujetos de estudio tenían evidencia de obstrucción al flujo aéreo, el deterioro es mucho peor en los que fumaban, lo que implica que incluso, el hábito de fumar de forma leve es particularmente peligroso para los pacientes con la enfermedad (40).
Año 1981
Durante un estudio del efecto de la plasmaféresis en el metabolismo glicofosfolípidos, un paciente con enfermedad de Fabry observó una notable mejoría en sus acroparestesias. Por lo cual se realizó un estudio controlado. Se realizaron observaciones de la conducción nerviosa, clasificados en pruebas de ejercicio y evaluaciones psicométricas, durante y después de dos series de tres bolsas de plasma: un verdadero intercambio de plasma y el otro un "simulacro" de control en el que la paciente recibió su propio plasma. Todos los observadores y el paciente eran cegados por unanimidad para disminuir beneficios para el procedimiento simulado. Este estudio demuestra la necesidad de más estudios en esta situación (41).
Año 1982
Sheth y Thomas publican los cambios en el electrocardiograma (ECG) de 47 miembros de tres familias con la enfermedad de Fabry. El grupo control con niveles de a-GAL A normales tenían ECG sin alteraciones. Diez de los 12 hombres afectados con niveles de a-Gal A menores del 10% de lo normal tenían signos de hipertrofia ventricular izquierda y cinco tenían cambios en el segmento onda ST y onda T. De las 16 mujeres heterocigotas, no había pruebas de crecimiento ventricular, pero había seis con anomalías de la conducción (42).
Por otra parte, se informa el caso de un paciente con enfermedad de Fabry de larga evolución, al cual se le realizó análisis bioquímico y patológico del hígado. Tanto los hepatocitos, así como los macrófagos periportales mostraron la acumulación de lípidos en forma de material amorfo a manera de pilas lamelares. Las inclusiones de lípidos a nivel de los macrófagos periportales eran mucho más grandes que en los hepatocitos. El análisis bioquímico mostró un aumento del contenido de 3-ceramida, con pequeñas concentraciones elevadas de otros glicofosfolípidos. La casi normal arquitectura hepática y la conservación de los organelos de los hepatocitos están de acuerdo con la observación de que la afectación hepática en la enfermedad de Fabry tiene mínima importancia clínica (43).
Año 1983
La hemodinámica periférica en las extremidades se estudió en ocho pacientes con la enfermedad de Fabry por medio de oclusión venosa segmentaria, neumopletismografía y sondas térmicas. Los resultados obtenidos fueron comparados con 10 sujetos normales. La resistencia vascular en el antebrazo en pacientes con Fabry fue significativamente mayor que en sujetos normales. La capacitancia venosa en antebrazo en los pacientes con la enfermedad fue significativamente menor. Las amplitudes del pulso no mostraron diferencias significativas en ningún segmento (parte superior del brazo, muñeca, muslo, por encima y por debajo de la rodilla y la pantorrilla) entre los dos grupos. La circulación, pulso y temperatura de los dedos fue menor en los pacientes con Fabry. Estos datos sugirieron la posibilidad latente de que una mayor descarga simpática adrenal, así como la acumulación de glicolípidos en el sistema nervioso autónomo y las paredes de los vasos (44).
Año 1986
Se publica un trabajo recopilando los ecocardiogramas de 35 pacientes con la enfermedad, 23 homicigotos y 12 heterocigotos para determinar si el compromiso cardiaco se podría detectar de manera no invasiva. Los resultados demostraron que los pacientes varones homocigotos tenían mayor diámetro de la raíz aórtica, grosor del séptum interventricular y una mayor masa ventricular que en las mujeres heterocigóticas. La masa ventricular izquierda por metro cuadrado de superficie corporal daba buena correlación con la gravedad de la enfermedad clínica, sugiriendo depósito progresivo de glicosfosfolípidos. Los heterocigotos mayores (más de 25 años) tenían hallazgos más destacados de la enfermedad cardiaca que los varones más jóvenes. A pesar de que el prolapso de la válvula mitral se identificó en 12 (54%) de 23 hombres homicigotos y en 7 (58%) de 12 de las mujeres heterocigotas, su presencia no se correlaciona con la gravedad de la enfermedad clínica. Por lo tanto, evidencia ecocardiográfica de la enfermedad de Fabry parece tener en este estudio relación con la edad, con la gravedad de la enfermedad y puede ser un marcador no invasivo útil para seguir la progresión y la posible regresión en caso que la terapia esté disponible (45).
Año 1987
Friedlaender y cols, presentan un interesante caso sobre los cambios clínicos y en la biopsia renal de un paciente ocho años después del trasplante de riñón. El injerto mantiene la función normal. Histología del injerto en microscopía de luz no mostró anomalías que recuerda a los enfermos de riñón nativo. La microscopía electrónica reveló ocasionales pequeñas figuras de mielina que estuvieron presentes sólo en el endotelio vascular (46).
Año 1989
Se realiza el primer informe histopatológico en pacientes con síntomas otológicos. Se describen las historias clínicas, audiometría e histología del hueso temporal de dos pacientes con este raro trastorno. Ambos pacientes demostraron pérdida auditiva neurosensorial bilateral. El oído medio presentaba derrame seropurulento y la mucosa era hiperplásica. El ligamento espiral y los giros eran atróficos con pérdida de vello, principalmente en las células basales de la gira. El número de células ganglionares espirales se redujo en todos los huesos temporales, sin embargo, la acumulación de glicosfosfolípidos no se observó en los ganglios de la espiral (47).
Año 1991
Von Scheidt y cols informan una variante de la enfermedad de Fabry aparentemente limitada a miocardio. Se trataba de un hombre de 54 años con angina de pecho inexplicable con hallazgos estructurales y funcionales dentro de los límites normales. Se realizó biopsia de endomiocardio evidenciando inclusiones lisosomales típicas, pero ningún otro hallazgo patológico o clínico fue encontrado. Posterior a esto se aisló y secuenció una cadena de DNA cuya secuencia genómica humana codificaba para a-Gal A. Aplicando estas técnicas se pudo evidenciar una mutación de este gen en el exón 6, el cual codifica para una enzima con actividad residual (48).
Año 1996
Por parte del departamento de medicina interna de la Universidad de Tokio, se presenta el caso de un paciente de 37 años con enfermedad de Fabry quien ingresó a urgencias por dolor torácico típico. Se realizó angiografía coronaria no encontrándose estenosis. Sin embargo, se presentó espasmo arterial después de la administración de acetilcolina. Se consideró que la injuria celular endotelial estaba asociada al depósito de glicofosfolípidos, considerándose como el principal mecanismo causal (49).
Año 1997
Se publica un estudio liderado por el Departamento de Genética del Instituto de Ciencias Médicas de Tokio, donde se aplica reacción en cadena de la polimerasa (PCR) simple o ligada a conformación de polimorfismo (SSCP) para la detección de mutaciones genéticas que causan la enfermedad de Fabry. Diecinueve de las 22 mutaciones conocidas mostraron cambios de movilidad electroforética en el análisis de PCR-SSCP. A continuación, el ADN de los pacientes diagnosticados con la forma clásica de la enfermedad fue sometido a análisis de PCR-SSCP y cuatro nuevas mutaciones y un polimorfismo neutro se identificaron, además, la identificación de un heterocigoto asintomático y un homocigoto con manifestaciones clínicas moderadas se ha logrado mediante la aplicación de este método a una familia con la variante de la enfermedad. Este trabajo muestra que la PCR- SSCP, derivada de fragmentos de DNA amplificado desnaturalizado, es útil para el diagnóstico genético de la enfermedad de Fabry de etiología heterogénea. Además, este método permite el diagnóstico temprano lo que contribuiría en prevenir el dolor y la trombosis (50).
Año 2000
Schiffmann y cols investigaron los efectos de la infusión de a-GaL A en 10 pacientes hombres con Fabry sin síntomas agudos. Las infusiones fueron bien toleradas en todos los pacientes. Se realizaron biopsias del tejido hepático en aproximadamente dos días después de la iniciación del manejo, encontrándose la enzima en varios tipos de células, incluidas las células endoteliales sinusoidales, células de Kupffer y los hepatocitos, lo que sugiere la difusión a través del receptor de manosa 6-fosfato. En tejido, la vida media en el hígado fue superior a 24 horas, que es mayor casi en un 50% en comparación con la plasmática. Después de la dosis única de alfa-GAL A, nueve de los 10 pacientes habían reducido significativamente niveles de globotriaosilceramida (Gb), tanto en el hígado como en las células epiteliales tubulares renales en el estudio del sedimento urinario. Estos datos demuestran que las infusiones de alfa-Gal A preparada a partir de fibroblastos humanos son seguras y bioquímicamente activas en pacientes con enfermedad de Fabry. El grado de reducción de sustrato visto en el estudio puede ser clínicamente significativo en vista del hecho de que la carga de Gb en estos pacientes aumenta gradualmente a lo largo de décadas. En conjunto, estos resultados sugieren que la enzima de reemplazo puede ser una terapia efectiva para los pacientes con este trastorno metabólico y podría lograrse esto con administración semanal o cada dos semanas (51).
Año 2001
En esta oportunidad Schiffmann y parte de su grupo publican un ensayo clínico doble ciego controlado y aleatorizado que buscaba evaluar la eficacia y seguridad de la infusión de a-Gal A administrada semanalmente por 12 dosis. Encontraron disminución de la severidad del dolor neuropático de manera estadísticamente significativa en comparación al grupo placebo. A nivel renal disminuyó el crecimiento mesangial glomerular de manera también significativa. En cuanto a la tasa de depuración de insulina, no hubo diferencias, pero sí en la depuración de creatinina. En los pacientes tratados con a-Gal A hubo reducción aproximada del 50% en los niveles plasmáticos de glicofosfolípidos, mejoría significativa en la conducción cardiaca, y un aumento significativo en el peso corporal. Este estudio corrobora con una metodología impecable la importancia del reemplazo enzimático a nivel multifuncional en los pacientes con Fabry (52).
Del mismo modo el International Collaborative Fabry Disease Study Group publica un estudio multicéntrico aleatorizado doble ciego, controlado con placebo, que tenía como objetivo también evaluar la seguridad de la a-Gal A recombinante en 58 pacientes tratados cada dos semanas por 20 semanas. Posterior a esto, todos los pacientes recibieron a-Gal A recombinante, en un estudio abierto de extensión. En el estudio doble ciego, 20 de los 29 pacientes del grupo que recibió a-Gal A no tenían depósitos endoteliales microvasculares de Gb después de 20 semanas, en comparación con ninguno de los 29 pacientes en el grupo placebo. Los pacientes con terapia enzimática de igual forma habían disminuido los depósitos de Gb en la piel y corazón de manera significativa. Los niveles plasmáticos de Gb se correlacionaron directamente con disminución de los depósitos microvasculares. Después de seis meses de tratamiento en la segunda parte el estudio, todos los pacientes del ex grupo placebo y 98% de los pacientes de la terapia enzimática habían tenido desaparición de los depósitos endoteliales microvasculares de Gb. La incidencia de la mayoría de los tratamientos relacionados con eventos adversos fue similar en los dos grupos, con excepción de leves a moderadas reacciones de perfusión (es decir, escalofríos y fiebre). Seroconversión de IgG se produjo en la mayoría de los pacientes que recibieron a-GAL A recombinante. Este trabajo orientó a que la terapia debe ser continuada para así reducir los depósitos de glicofosfolípidos en otros tipos de células donde los niveles de la enzima es menor (53).
Año 2002
Thurberg y cols publican un estudio doble ciego, aleatorizado, controlado con placebo seguido de un periodo de seis meses de estudio abierto de extensión utilizando la enzima humana recombinante, alfa - Gal A (r-halphaGal A) administrada cada dos semanas. Las evaluaciones revelaron acumulaciones de Gb en casi todos los tipos de células renales vasculares incluyendo células endoteliales, células del músculo liso vascular, células mesangiales y células intersticiales, en particular con acumulaciones densas en los podocitos y en las células epiteliales tubulares distales. Después de 11 meses de tratamiento de r-halphaGal A se completa desaparición de glicolípidos del endotelio de los vasos, así como de las células mesangiales del glomérulo y células intersticiales de la corteza. Se observó moderada disminución en las células musculares lisas de pequeñas arterias y arteriolas. El epitelio tubular distal también demostró evidencia de disminución, aunque más limitada que la observada en otros tipos de células. Esto corrobora lo demostrado en el 2002, en cuanto a las ventajas de la terapia enzimática a largo plazo, en este caso invirtiendo la posibilidad de falla renal (54).
Año 2003
Se presenta un ensayo que quería corroborar la eficacia de la terapia de reemplazo enzimático con el placebo sobre las puntuaciones de dolor neuropático fuera de los medicamentos para el dolor y utilizando las pruebas sensoriales cuantitativas, prueba reflejo axón sudomotor (QSART), test del sudor y de la termorregulación. Se demostró una modesta, pero significativa mejoría en las manifestaciones clínicas de neuropatía de la pequeña fibra. De igual forma, se postula el QSART como herramienta para optimizar la dosis y la frecuencia de la terapia de reemplazo enzimático (55).
Por parte del Departamento de Medicina Interna y Reumatología de la Universidad Autónoma de Madrid se publica el caso de una paciente de 45 años con diagnóstico de Fabry, que inicialmente presentaba dolores recurrentes en extremidades debidos a la enfermedad y no presentaba signos inflamatorios ni erosiones en las radiografías. Sin embargo, en la evolución posterior se puso de manifiesto un cambio en la sintomatología, apareciendo rigidez y signos inflamatorios en la exploración física de las articulaciones, por lo que realizó un estudio inmunológico y radiológico y se diagnosticó artritis reumatoide. Este es el primer caso de asociación de enfermedad de Fabry y artritis reumatoide. Porque aunque sí existe afectación articular en la enfermedad de Fabry por depósito de lípidos en la piel y en los compartimentos sinoviales con alargamiento de la cápsula articular y los tendones, hay otros cambios como osificación de las entesis en la inserción de las estructuras fibrosas y algunas erosiones intra y extraarticulares. Estas erosiones son infrecuentes y se diferencian de las que aparecen en la artritis reumatoide, en la que son yuxtaarticulares (56).
Año 2004
Se publican los resultados del FOS (Fabry Outcome Survey) que combina los datos de las clínicas europeas sobre la historia natural de esta enfermedad y además supervisa la eficacia y seguridad del tratamiento a largo plazo. Se basó en una cohorte de 366 pacientes de 11 países. Se encontró que el diagnóstico erróneo de la enfermedad era común, y la media de retraso de la aparición de los síntomas al diagnóstico fue de 13,7 y 16,3 años en varones y mujeres, respectivamente. Aunque anteriormente se pensaba que sólo los hombres homocigotos tenían manifestaciones graves, el FOS encontró que las mujeres heterocigotas también podrían tenerlas. Además, los signos y síntomas de la enfermedad de Fabry pueden estar presentes desde la primera infancia. Todo lo anterior abrió el camino para el espectro de la terapia de reemplazo enzimático (57).
Año 2005
Por parte de la Escuela de Medicina de Nippon se recopilan las manifestaciones cardiacas de la enfermedad de Fabry. Se incluye la hipertrofia ventricular izquierda usualmente con fracción de eyección conservada pero con moderada disfunción diastólica, con enfermedad coronaria estenótica o vasoespástica causante de angina de pecho. En cuanto a la insuficiencia valvular, se presenta en la mayor parte en la mitral, causada por la acumulación de Gb. Esta última es también la causante de bloqueo A-V, PR corto, arritmias y muerte súbita. Las imágenes doppler permiten realizar diagnóstico preclínico de la cardiomiopatía. Se da importancia a la terapia de reemplazo enzimático con disminución en los cambios estructurales en seguimiento imagenológico. No obstante se pone de manifiesto el inconveniente de la terapia de reemplazo, por la vida media corta de las moléculas por lo que es requerido un largo periodo de tiempo; por esto, se da importancia a la terapia enzimática mediada por genes (58).
Año 2006
Se publica un trabajo que tenía como objetivo evaluar la seguridad y la eficacia de tratamiento con la enzima alfa agalsidasa en pacientes pediátricos con enfermedad de Fabry. Se realizó a través de un estudio abierto por periodo de seis meses en 24 niños (19 varones y cinco niñas) con una edad media de 11,8 años. Se encontró adecuada tolerancia de la terapia enzimática. En seis niños y una niña reacciones moderadas durante la infusión. Un niño desarrolló anticuerpos contra la alfa agalsidasa. Los niños mostraron una reducción significativa de los niveles plasmáticos de Gb. La tasa de filtración glomerular estimada, la estructura y función cardiaca fueron normales y no cambiaron en un periodo de 26 semanas. Todos los índices de variabilidad del ritmo cardiaco mejoraron significativamente en los niños. Hubo disminución de analgésicos de manera importante en el caso de neuropatía concomitante. Aunque este estudio tiene limitaciones metodológicas, proporciona importantes aportes en el futuro de la terapia en niños (59).
Brent y Erica Kelly informan un caso de angiokeratoma corporis difussum el cual no se asoció a deficiencia enzimática, ni anormalidades subsecuentes sistémicas. Se trataba de un hombre de 33 años quien presenta pápulas rojo-púrpuras en la ingle, las cuales han aumentado de tamaño desde la pubertad. Las lesiones iniciaron en la niñez en codos y rodillas. La biopsia reveló ortokeratosis, acantosis epidérmica y dilatación superficial de capilares. La microscopía electrónica mostró células endoteliales normales con lisosomas no laminados o aumentados de tamaño. Análisis enzimático de a-D-galactosidasa, a- N- acetilgalactosaminidasa, a-D-manosidasa y a-L-fucosidasa se encontraban dentro de los límites normales. Este es el décimo caso informado en la literatura, cuya fisiopatología no es conocida, pero se considera una entidad distinta con un curso benigno. En estos casos la terapia láser genera buenos resultados (60).
Año 2007
Ries y colaboradores realizan un estudio retrospectivo con el fin de evaluar la pérdida de la audición en pacientes con enfermedad de Fabry, remitidos al Instituto de Salud de Bethesda. Se evaluaron 109 pacientes incluyendo hombres y mujeres, a los cuales se les realizó evaluación audiológica completa. En los pacientes con mayor pérdida auditiva se documentaba de forma concomitante lesiones en sustancia blanca, neuropatía periférica y pérdida de la función renal. De igual manera la deficiencia de a- Gal A determinaba mal pronóstico auditivo (61).
Teniendo en cuenta la alta prevalencia de dolor en los pacientes con Fabry, el grupo europeo FOS publica un estudio retrospectivo con el fin de evaluar la naturaleza y prevalencia del dolor en una cohorte de pacientes y de forma concomitante evaluar el efecto de la terapia de reemplazo enzimático en el mismo. Encontraron que la prevalencia de dolor era de 81,4% en hombres y 65,3% en mujeres siendo más frecuente en manos y pies, afectando la actividad diaria de manera más representativa en mujeres. El 58% de los pacientes que recibieron terapia con a agalsidasa, presentaron mejoría del dolor de manera significativa a los 24 y 36 meses (62).
Año 2008
Considerando la respuesta terapéutica variable de las preparaciones enzimáticas, Vedder y colaboradores, estudiaron la medida de anticuerpos contra a-Gal, excreción urinaria, niveles plasmáticos de Gb y citotriosidasa en 52 pacientes después de 12 meses de tratamiento con alfa agalsidasa o ß agalsidasa. Encontraron que los anticuerpos a-Gal eran desarrollados con mayor frecuencia en los hombres y esto interfiere en la excreción de Gb. Las infusiones a 1 mg/kg reducían con mayor eficacia la Gb, existía mejor estabilidad de la función renal y en la reducción de la masa del ventrículo izquierdo. Este trabajo plantea hipótesis sobre el valor de los anticuerpos en el curso clínico y su posible relación con el genotipo y la presencia de proteínas enzimáticas (63).
Nasi Samiy publica una excelente revisión sobre el compromiso ocular de la enfermedad. Se ratifica a las opacidades corneales como la alteración más común. En el 95% de los hombres y 88% de las mujeres con los genes portadores, las lesiones se ubican en las capas epiteliales y subepiteliales de la córnea o cerca a la membrana de Bowman. Las lesiones comienzan en la niñez con informes desde los 2 y 6 años. En cuanto a la córnea verticilata con una frecuencia entre 53% a 94% es más frecuente en mujeres, y es llamada por muchos autores como "distrofia corneal", "keropatia vortex" o "vortex de Fleischer". Por otra parte, las anormalidades vasculares conjuntivales son más comunes en hombres. Las opacidades lenticulares las cuales son visibles a través de retroiluminación son encontrados en 70% de los hombres y 35% de las mujeres. Las cataratas también son más frecuentes en hombres y se caracterizan por ser bilaterales. El compromiso en retina es dado por tortuosidad vascular y es encontrado en el 77% de los hombres y 13% de las mujeres (64).
Año 2009
Se publica un trabajo que tenía como fin evaluar si anti-IgG alfa -Gal interfería con la eficacia terapéutica del reemplazo enzimático; se realizaron análisis retrospectivos usando datos obtenidos de un total de 134 adultos, hombres y mujeres con enfermedad de Fabry que fueron tratados con beta agalsidasa. Los análisis no revelaron una correlación entre los alfa-Gal contra los títulos de IgG y la aparición de eventos clínicos o la tasa de cambio en la estimación de tasa de filtración glomerular durante el tratamiento. No se encontró significancia estadística entre los niveles de IgG y elevaciones anormales de Gb. Sin embargo, sí se encontró significancia estadística entre la elevación de los anticuerpos y los niveles de Gb en las células endoteliales capilares de la piel. Estos resultados orientarían al seguimiento de anticuerpos durante la terapia de reemplazo enzimático (65).
Agradecimientos
A Jaime Muñoz Carreño por su contribución en la edición final de este trabajo.
Declaración de conflicto de intereses
No existe conflicto de intereses entre los autores, las instituciones y el contenido del presente trabajo.
Fuente de financiación
Declaramos que para la elaboración de este manuscrito no se ha contado con ninguna fuente de financiación.
Referencias
1. Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004; 34: 236-42. [ Links ]
2. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999; 281: 249-54. [ Links ]
3. Eng CM, Germain DP, Banikazemi M, Warnock DG, Wanner C, Hopkin RJ, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med 2006; 8: 539–48. [ Links ]
4. Breunig F, Wanner C. Update on Fabry disease: kidney involvement, renal progression and enzyme replacement therapy. J Nephrol 2008; 21: 32-7. [ Links ]
5. Fabry H. An historical overview of Fabry disease. J Inherit Metab Dis 2001; 24: 3–7. [ Links ]
6. Anderson W. A case of angeio-keratoma. Brit J Dermatol 1898; 10: 113-7. [ Links ]
7. Fabry J. Ein Beitrag zur Kenntnis der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae). Arch Dermatol Syph 1898; 43: 187-200. [ Links ]
8. Fabry J. Archiv f. Derm. u. Syph. Bd. CXXIII 294-308 [ Links ]
9. Steiner L, Voerner H. Angiokeratoma miliare, eine ideiopathische Gefaesserkrankung . Deutsch Arch klin Med 1909; 96: 105. [ Links ]
10. Weicksel J. Angiomatosis bezw, Angiokeratoma universalis. Deutsch Med Woschr 1925; 51: 898-900. [ Links ]
11. Ruiter M, Pompen AWM. Angiokeratoma corporis diffusum (universale) mit kardiovasorenale m Symptomenkomplex bei 3. Brüdern. Arch Derm Syph 1939; 179: 169. [ Links ]
12. Ruiter M, Pompen AWM, Wijers HJG. Uber internmedizinische und pathologisch-anatomisch e Befunde bei Angiokeratome corporis diffusum (Fabry). Dermatol 1947; 94: 1. [ Links ]
13. Scriba K, Zur Pathogenes. Des Angiokeratoma corporis diffusum. Fabry mit cardio-vasorenale m Symptomenkomplex Verhandlg. Deutsch Path Gesellsch 1950; 34: 221. [ Links ]
14. Hornboste l H, Spier W, Koch H. Angiokeratoma corporis diffusum universale (Fabry) mit kardiorenalem Symptomenkomplex als allgemeine Erkrankung. Arztl Woschr. 1951; 49. [ Links ]
15. Fessas P, Wintrobe MM, Cartwright GE. Angiokeratoma corporis diffusum: First American Report of a Rare Disorder. AMA Arch Intern Med 1955; 95: 469-81. [ Links ]
16. Witschel H, Meyer W. Der Morbus Fabry als Beispiel einer erblichen Lipoidspeicherkrankheit. Neuere Gesiehtspunkte zur Pathogenese, Klinik und Morphologie. Klin Wschr 1968; 46: 72-6. [ Links ]
17. Ruiter M. Angiokeratoma corporis diffusum Syndrome and its cutaneous manifestations: review and personal experience in the last ten years. Hautarzt 1958; 9: 15-9. [ Links ]
18. Colley JR, Miller DL, Hutt MSR, Wallace HJ, Wardener HE. The renal lesion in angiokeratoma corporis diffusum. Br Med J 1958; 1: 1226-8. [ Links ]
19. Karr WJ Jr. Fabry's disease (angiokeratom a corporis diffusum universale). An unusual syndrome with multisystem involvement and unique skin manifestations. Am J Med 1959; 27: 829–35. [ Links ]
20. Hofmann A, Hauser W. Angiokeratoma corporis diffusum (Fabry) mit cerebralen Manifestationen. Dtsch Z Nervenheilkd 1962;183: 351-62. [ Links ]
21. Sweeley CC, Klionsky B. Fabry's disease: Classification as a sphingolipidosis and partial characterization of a novel glycolipid. J Biol Chem 1963; 238: 3148–50. [ Links ]
22. deGroot WP. Angiokeratoma corporis diffusum Fabry. Dermatologica 1964; 128: 321–49. [ Links ]
23. Opitz JM, Stiles FC, Wise D, Race RR, Sanger R, Gemmingen GR, et al. The Genetics of Angiokeratoma Corporis Diffusum (Fabry's Disease) and Its Linkage Relations with the Xg Locus. Am J Hum Genet 1965; 17: 325-42. [ Links ]
24. Hashimoto K, Gross BG, Lever WF. Angiokeratoma corporis diffusum (Fabry). Histochemical and electron microscopical studies of the skin. J Invest Dermatol 1965; 44: 119–28. [ Links ]
25. Dempsey H, Hartley MW, Carroll J, Balint J, Miller RE, Frommeyer WB Jr. Fabry's disease (angiokeratom a corporis diffusum): case report on a rare disease. Ann Intern Med 1965; 63: 1059–68. [ Links ]
26. Brady RO, Gal AE, Bradley RM, Martenson E, Warshaw AL, Laster L. Enzymatic defect in Fabry's disease: ceramidetri hexosidase deciency. N Engl J Med 1967; 276: 1163–7. [ Links ]
27. Rae AI, Lee JC, Hopper J Jr. Clinical and electron microscopic studies of a case of glycolipid lipoidosis (Fabry's disease). J clin Path 1967; 20: 21-7. [ Links ]
28. Philippart M, Sarlieve L, Manacorda A. Urinary glycolipids in Fabry's disease. Their examination in the detection of atypical variants and the pre-symptomatic state. Pediatrics 1969; 43: 201–6. [ Links ]
29. Brady RO, Uhlendorf BW, Jacobson CB. Fabry's disease: antenatal detection. Science 1971; 172: 174–5. [ Links ]
30. Desnick RJ, Sweeley CC. Prenatal defect of Fabry's disease. In: Dorfman A, ed. Antenatal diagnosis. Chicago, IL: University of Chicago Press, 1971.p.185. [ Links ]
31. Desnick RJ, Allen KY, Simmons RL, Woods JE, Anderson CF, Najarian JS, et al. Correction of enzymatic de ciencies by renal transplantation: Fabry's disease. Surgery 1972; 72: 203–11. [ Links ]
32. Pilz H, Volles E, Paul HA, Denden A. Neurologische Symptome bei Fabryscher Krankheit (Angiokeratoma corporis diffusum). J Neurol 1972; 202: 307-22. [ Links ]
33. Brady RO, Tallman JF, Johnson WG, Gal AE, Leahy WR, Quirk JM, et al. Replacement therapy for inherited enzyme deciency. Use of puried ceramidetrihexosidase in Fabry's disease. N Engl J Med 1973; 289: 9–14. [ Links ]
34. Mckusick VA. Genetics and dermatology or if I were to rewrite Cockayne's inherited abnormalities of the skin. J Invest Dermatol 1973; 60: 343-59. [ Links ]
35. Roth J, Schulze E, Raabe G, Waldmann G. Analytische Studie des Morbus Fabry. Virchows Archiv A Path Anat Histol 1974; 363: 287-301. [ Links ]
36. Pierides AM, Holti G, Crombie AL, Roberts DF, Gardiner SE, Colling A, et al. Study on a family with Anderson–Fabry's disease and associated familial spastic paraplegia. J Med Genet 1976; 13: 455–61. [ Links ]
37. Johnson DL, Desnick RJ. Molecular pathology of Fabry's disease. Physical and kinetic properties of alpha-galactosidase A in cultured human endothelial cells. Biochim Biophys Acta 1978; 538:195-204. [ Links ]
38. Sher NA, Letson RD, Desnick RJ. The ocular manifestations in Fabry's disease. Arch Ophthalmol 1979; 97: 671-6. [ Links ]
39. Pyeritz RE, Ullman MD, Moser AB, Braine HG, Moser HW. Plasma exchange removes glycosphingolipid in Fabry disease. Am J Med Genet 1980; 7: 301-7. [ Links ]
40. Rosenberg DM, Ferrans VJ, Fulmer JD, Line BR, Barranger JA, Brady RO, et al. Chronic airflow obstruction in Fabry's disease. Am J Med 1980; 68: 898-905. [ Links ]
41. Braine HG, Pyeritz RE, Folstein MF, Moser HB, Ullman MD. A prospective double-blind study of plasma exchange therapy for the acroparesthesia of Fabry's disease. Transfusion 1981; 21: 686-9. [ Links ]
42. Sheth KJ, Thomas JP Jr. Electrocardiograms in Fabry's disease. J Electrocardiol 1982; 15:153-6. [ Links ]
43. Meuwissen SG, Dingemans KP, Strijland A, Tager JM, Ooms BC. Ultrastructural and biochemical liver analyses in Fabry's disease. Hepatology 1982; 2: 263-8. [ Links ]
44. Seino Y, Vyden JK, Philippart M, Rose HB, Nagasawa K. Peripheral hemodynamics in patients with Fabry's disease. Am Heart J 1983; 105: 783-7. [ Links ]
45. Goldman ME, Cantor R, Schwartz MF, Baker M, Desnick RJ. Echocardiographic abnormalities and disease severity in Fabry's disease. J Am Coll Cardiol 1986; 7:1157-61. [ Links ]
46. Friedlaender MM, Kopolovic J, Rubinger D, Silver J, Drukker A, Ben-Gershon Z, et al. Renal biopsy in Fabry's disease eight years after successful renal transplantation. Clin Nephrol. 1987; 27: 206-11. [ Links ]
47. Schachern PA, Shea DA, Paparella MM, Yoon TH. Otologic histopathology of Fabry's disease. Ann Otol Rhinol Laryngol 1989; 98:359-63. [ Links ]
48. von Scheidt W, Eng CM, Fitzmaurice TF, Erdmann E, Hübner G, Olsen EG, et al. An atypical variant of Fabry's disease with manifestations confined to the myocardium. N Engl J Med 1991; 324: 395-9. [ Links ]
49. Ogawa T, Kawai M, Matsui T, Seo A, Aizawa O, Hongo K, et al. Vasospastic angina in a patient with Fabry's disease who showed normal coronary angiographic findings. Jpn Circ J 1996; 60: 315-8. [ Links ]
50. Takata T, Okumiya T, Hayashibe H, Shimmoto M, Kase R, Itoh K, et al. Screening and detection of gene mutations in Japanese patients with Fabry disease by non-radioactive single-stranded conformation polymorphism analysis. Brain Dev 1997; 19: 111-6. [ Links ]
51. Schiffmann R, Murray GJ, Treco D, Daniel P, Sellos-Moura M, Myers M, et al. Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci U S A 2000; 97: 365-70. [ Links ]
52. Schiffmann R, Kopp JB, Austin HA 3rd, Sabnis S, Moore DF, Weibel T, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001; 285: 2743-9. [ Links ]
53. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, et al. Safety and efficacy of recombinant human alpha-galactosidase A--replacement therapy in Fabry's disease. N Engl J Med 2001; 345: 9-16. [ Links ]
54. Thurberg BL, Rennke H, Colvin RB, Dikman S, Gordon RE, Collins AB, et al. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int 2002; 62: 1933-46. [ Links ]
55. Schiffmann R, Floeter MK, Dambrosia JM, Gupta S, Moore DF, Sharabi Y, et al. Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve 2003; 28: 703-10. [ Links ]
56. Arias N, Barbado F, Pérez G, Pérez C, Casal V, Vázquez J. Enfermedad de Fabry asociada a artritis reumatoide. Una encrucijada multisistémica. An Med Interna (Madrid) 2003; 20: 28-30. [ Links ]
57. Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest 2004; 34: 236-42. [ Links ]
58. Seino Y, Takahashi H, Fukumoto H, Utsumi Kouichi, Hirai Y. Cardiovascular manifestations of Fabry disease and the novel therapeutic strategies. J Nippon Med Sch 2005; 72: 254-61. [ Links ]
59. Ries M, Clarke JT, Whybra C, Timmons M, Robinson C, Schlaggar BL, et al. Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics 2006; 118: 924-32. [ Links ]
60. Kelly B, Kelly E. Angiokeratoma corporis diffusum in a catient with no recognizable enzyme abnormalities. Arch Dermatol 2006; 142: 615-8. [ Links ]
61. Ries M, Kim HJ, Zalewski CK, Mastroianni MA, Moore DF, Brady RO, et al. Neuropathic and cerebrovascular correlates of hearing loss in Fabry disease. Brain 2007; 130: 143-50. [ Links ]
62. Hoffmann B, Beck B, Sunder-Plassmann G, Borsini W, Ricci R, Mehta A. Nature and Prevalence of Pain in Fabry Disease and Its Response to Enzyme Replacement TherapyA retrospective analysis from the Fabry Outcome Survey. Clin J Pain 2007; 23: 535–42. [ Links ]
63. Vedder AC, Breunig F, Donker-Koopman WE, Mills K, Young E, Winchester B, et al. Treatment of Fabry disease with different dosing regimens of agalsidase: Effects on antibody formation and GL-3. Mol Genet Metab 2008; 94: 319–25. [ Links ]
64. Samiy N. Ocular features of Fabry disease: diagnosis of a treatable life-threatening disorder. Surv Ophthalmol 2008; 53: 416-23. [ Links ]
65. Bénichou B, Goyal S, Sung C, Norfleet AM, O'Brien F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol Genet Metab 2009; 96:4-12. [ Links ]

