










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.35 no.1 Bogotá Jan./Mar. 2010
PRESENTACIÓN DE CASOS
Devic's Disease (Neuromyelitis optica)
Alfredo Pinzón(1), Tatiana Echeverry(2),
Aida Bibiana Rodríguez(3) • Bogotá, D.C.
(1) Especialista en Medicina Interna, Hospital Universitario de La Samaritana; Profesor Clínico Universidades Javeriana y La Sabana;
(2) Residente de Medicina Interna, Hospital Universitario de La Samaritana - Universidad de La Sabana;
(3) Servicio Social Obligatorio, (Medicina Interna), Hospital Universitario de La Samaritana.
Correspondencia. Carrera 8 No. 0-55 sur, Bogotá, D.C. (Colombia).
E-mail: Alfredo.Pinzon@hus.org.co
Recibido: 31/III/09 Aceptado: 17/XII/09
Resumen
Presentamos el caso de una mujer joven inicialmente tratada por esclerosis múltiple, quien presentó recurrencia con compromiso ocular severo. La enfermedad de Devic es un desorden desmielinizante que se presenta como una mielitis transversa asociada con neuritis óptica, típicamente bilateral. El principal diagnóstico diferencial es justamente la esclerosis múltiple (Acta Med Colomb 2010; 35: 21-25).
Palabras clave: neuromielitis óptica, enfermedad de Devic, esclerosis múltiple.
Abstract
We present a case report about a young woman initially treated as having multiple sclerosis, who relapsed with serious visual impairment. Devic's disease is a demyelinating disorder that presents as transverse myelitis associated with optic neuritis, typically bilateral. Multiple sclerosis is in fact the main differential diagnosis (Acta Med Colomb 2010; 35: 21-25).
Key words: neuromyelitis optica, Devic's disease, multiple sclerosis.
Descripción del caso
Mujer de 28 años, natural y procedente de Bogotá, ama de casa, quien consultó por cuadro clínico de una semana de evolución consistente en debilidad progresiva de miembros superiores e inferiores hasta imposibilidad total para el movimiento y la marcha, asociada a disnea y dolor en región cervical. A la revisión por sistemas refería fiebre no cuantificada, disuria y poliuria. Los antecedentes positivos eran: esclerosis múltiple diagnosticada 14 meses atrás mediante resonancia magnética cervicotoracolumbar que fue reportada como: lesión hiperintensa única en secuencia T2 localizada en la médula cervical (comprometiendo tres cuerpos vertebrales), que podría estar en relación con mielitis transversa aguda idiopática y como segunda posibilidad esclerosis múltiple, con abombamiento universal de la región medular cervical; en aquel momento la resonancia cerebral fue normal; episodios recurrentes de infección urinaria por vejiga neurogénica. Por estos motivos había sido hospitalizada en cinco ocasiones. Además, cesárea por parto gemelar que requirió manejo en UCI por eclampsia 15 meses antes de la enfermedad actual. Venía recibiendo amitriptilina 25 mg/día, propranolol 40 mg/ día y norfloxacina 400 mg cada 12 horas.
Al examen físico se encontró deshidratada, polipneica, con tensión arterial 130/60 mm Hg, frecuencia cardiaca 130 x min, frecuencia respiratoria 26 x min y temperatura axilar 36.5°C. cuello sin adenopatías. En la auscultación cardiopulmonar: ruidos cardiacos rítmicos sin soplos; ruidos respiratorios con murmullo vesicular simétrico sin agregados pulmonares. El abdomen estaba blando, con ruidos intestinales normales, sin masas ni visceromegalias. Las extremidades presentaban buena perfusión distal, con pulsos simétricos, sin edema.
Al examen neurológico alerta, orientada, sin déficit en la esfera mental. Pares craneanos motores normales. La fundoscopia mostró atrofia óptica en ojo izquierdo y presentaba pupilas dilatadas no reactivas (pupilas de Marcus Gunn). Se documentó cuadriparesia simétrica 1/5, sin déficit de la sensibilidad táctil. No se encontró alteración de los reflejos tendinomusculares. Signo de Hoffman positivo izquierdo y signo de Babinski bilateral.
Los exámenes paraclínicos mostraron:
Hemograma: leucocitos 5.810/mm3 (neutrófilos 78% y linfocitos 17%); hematocrito 36.4% con hemoglobina de 12 g/dL; plaquetas 229.000/fL.
Química sanguínea: nitrógeno ureico 13.02 mg/dL; creatinina sérica 0.46 mg/dL; glucemia 71 mg/dL; natremia 137 mEq/L; kaliemia 4.3 mEq/L; cloremia 104.88 mEq/L y magnesemia 2.1 mmol/L.
Uroanálisis: leucocitos >100/c, bacterias +++, nitritos positivo.
Factor Reumatoide: negativo.
Elisa para VIH: no reactivo. Estudios inmunológicos: ANAS y ENAS negativos. Radiografía de tórax: normal.Líquido cefalorraquídeo: incoloro, transparente, hematíes 12-15/c, leucocitos 7/mm3, glucosa 52 mg/dL y proteínas 17 g/L.
Resonancia magnética cervical: realce focal a nivel de la unión bulborraquídea tras la administración del medio de contraste que podría estar en relación con esclerosis múltiple; pequeño foco hiperintenso localizado en la sustancia blanca del centro semioval anterior izquierdo de características inespecíficas (Figura 1).
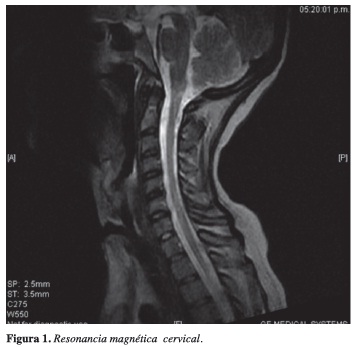
Evolución
Bajo la presunción diagnóstica de esclerosis múltiple se inició manejo con bolos de metilprednisolona y metotrexate, así como manejo antibiótico con ampicilina-sulbactam para la infección urinaria. Sin embargo, evolucionó tórpidamente, con deterioro del patrón respiratorio, hipercapnia e inminencia de falla respiratoria, por lo que fue trasladada a Unidad de Cuidados Intensivos para soporte ventilatorio mecánico. Allí se continuó manejo con gamma-globulina humana y azatioprina, requiriendo traqueostomía por intubación prolongada y gastrostomía. Recibió además ciclos antibióticos con piperacilina-tazobactam y meropenem por sendas infecciones nosocomiales. Después de los bolos de esteroide parenteral se continuó con prednisona oral. Persistió con amaurosis bilateral y cuadriparesia fluctuante 1/5 a 2/5, además de áreas de hipoestesia en parches, sin alteración de otros pares craneanos ni reflejos.
Fue valorada por oftalmología quienes encuentran atrofia óptica bilateral del 100% (Figura 2).
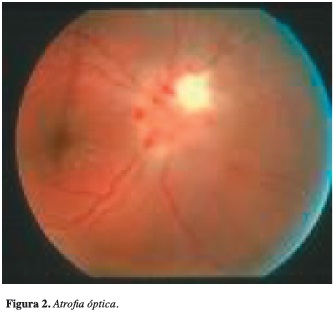
Además, presentó cuadro de disfonía posextubación por lo que se realizó nasofibrolaringoscopia que mostró estenosis subglótica del 70% de la luz. Igualmente, en una TAC de cuello se documentó trombosis de vena yugular derecha que fue confirmada mediante ultrasonido Doppler de cuello que reportó vena yugular derecha con trombo antiguo moderadamente hipogénico que no permitía colapso de la vena; por este motivo se inició anticoagulación terapéutica a largo plazo. Se continuó el manejo de rehabilitación y cuidados de traqueostomía.
Desafortunadamente, no se obtuvo recuperación del déficit visual y la fuerza muscular sólo mejoró a 3/5. Se prescribió manejo ambulatorio con azatioprina 100 mg/día, prednisona 35 mg/día, metoprolol 25 mg cada 12 horas, trazodone 50 mg/día y warfarina a dosis ajustada para INR terapéutico de 2,0 a 3,0.
Discusión
La neuromielitis óptica (NMO), también conocida como enfermedad o síndrome de Devic, fue descrita originalmente por el médico francés Eugene Devic en 1894, de quien deriva su epónimo (1). Es una enfermedad monofásica o recurrente, que se presenta en forma aguda como mielitis transversa más compromiso del nervio óptico bilateral. En 1999 se añadió a la definición las "recaídas", y se tomaron en cuenta para el diagnóstico los criterios del líquido cefalorraquídeo y los hallazgos de la resonancia magnética. En el líquido cefaloraquídeo hay pleocitosis con predominio de neutrófilos (a diferencia del predominio linfocitario visto en la esclerosis múltiple) y las bandas oligoclonales son poco frecuentes (15%). En 2004 se describieron los anticuerpos IgG contra aquaporina 4 como parte importante de la fisiopatología de la enfermedad (2, 3). La NMO tiene una distribución mundial, es más frecuente en mujeres (80%) que en hombres, y afecta a jóvenes o adultos entre 30 años y 40 años, presentando mayor incidencia en americanos, asiáticos, caucásicos, africanos y mediterráneos. Suele asociarse a procesos autoinmunes como lupus eritematoso sistémico, tiroiditis de Hashimoto, síndrome de Sjögren (10%-40%), o infecciosos como tuberculosis (4-6).
En la NMO hay múltiples ataques uni o bilaterales de neuritis óptica y los episodios de neuritis y mielitis casi siempre están separados por semanas, meses o años. Característicamente no afecta la sustancia blanca ni produce síntomas encefálicos, pero la afección de la columna cervical puede producir náuseas, hipo o incluso falla respiratoria, la que conduce a pobre pronóstico y casi siempre es la causa de la muerte en estos pacientes.
Por muchos años se asumió erróneamente que la mielitis transversa y la neuritis óptica debían ser simultáneas, y que la primera debía ser completa y la segunda bilateral (2). La NMO era considerada una enfermedad muy rara y durante el siglo XX hubo debate sobre si se trataba de una entidad nosológica diferente o si por el contrario era tan sólo una forma de esclerosis múltiple (EM). En el año 2005 fue posible distinguirla de esta última con base en su perfil clínico, imaginológico, serológico e inmunopatológico particular (7). Esta distinción fue necesaria para la implementación de tratamientos adecuados, ya que la forma recurrente de la enfermedad conlleva morbilidad y mortalidad significativas. El proceso agudo que afecta los nervios ópticos y la médula espinal puede ser simultáneo o estar separado por varias semanas. El cuadro clínico consiste en pérdida de la agudeza visual y mielitis con cuadriparesia o paraparesia, retención urinaria, espasmos y dolor en la región cervical y/o lumbar; la mielitis puede ser parcial o total, pero el criterio diagnóstico exige que en la resonancia magnética se encuentre compromiso de por lo menos tres cuerpos vertebrales (hipointensidades en T1 e hiperintensidades en T2) (1, 8). Por lo general, la resonancia cerebral será normal, aunque ocasionalmente pueden encontrarse lesiones hipotalámicas (hasta 10% de los pacientes) (4, 9, 10).
La enfermedad puede ser monofásica o recurrente. La primera ocurre en el 25% de los casos mientras que la segunda es la más frecuente (75%). El género femenino presenta mayor compromiso motor durante los ataques de mielitis. La forma recurrente de NMO afecta principalmente mujeres, con inicio que varía desde la infancia hasta adultos en sus cuarenta años o ancianos (5,11).
La presencia de un marcador sérico altamente específico (el autoanticuerpo llamado NMO-IgG) diferencia la NMO de la EM y ha ayudado a definir el espectro de desórdenes relacionados con NMO. El NMO-IgG reacciona específicamente contra los canales de agua acuaporina 4 (AQP4), que se encuentran principalmente a nivel de los astrocitos y la sustancia periacueductal, mientras que no están presentes en la mielina ni en los oligodendrocitos.
Los experimentos preliminares mostraron que el NMOIgG puede modular la función del AQP4 además de fijar el complemento (12-15). Los datos sugieren que estos autoanticuerpos derivados de las células B periféricas causan la activación del complemento, la desmielinización inflamatoria y la necrosis vista en la NMO. A diferencia de la EM, en la NMO el ataque no es mediado por células T sino por células B. Algunas características clínicas, imaginológicas y serológicas permiten diferenciar las dos entidades (Tabla 1) (4). La NMO puede manifestarse con marcadores séricos de autoinmunidad, como ANAs (hasta 50% de los casos), factor reumatoide y tiroglobulina positivos (3,16).
Aunque inicialmente se consideró que la enfermedad de Devic podría corresponder a una variedad de esclerosis múltiple (9), actualmente se acepta que es una entidad totalmente diferente (7).
Los criterios de Wingerchuk (1999) son útiles para establecer el diagnóstico de la enfermedad (8,12,17). Se dividen en absolutos y complementarios; a su vez, estos últimos pueden ser mayores y menores.
Criterios absolutos
- Neuritis óptica.
- Mielitis aguda.
- Ausencia de enfermedad por fuera del nervio óptico y la médula espinal.
Criterios complementarios mayores
- RM cerebral negativa.
- RM médula espinal con lesiones que comprometen tres o más cuerpos vertebrales.
- LCR con pleocitosis (≥50 leucocitos o cinco neutrófilos/ mm3).
Criterios complementarios menores
- Neuritis óptica bilateral.
- Neuritis óptica severa (agudeza visual <20/200 en al menos un ojo).
- Severa debilidad de más de una extremidad ≤ 2/5. Recientemente (2006), el mismo autor propuso una revisión de los criterios diagnósticos de la NMO definitiva (13, 18), a saber:
- Neuritis óptica.
- Mielitis aguda.
- Al menos dos de tres criterios de soporte: RM médula espinal con lesiones que comprometen tres o más cuerpos vertebrales; RM cerebral que no cumpla criterios de EM; serología positiva para el NMO-IgG.
También es útil la clasificación de Mandler (2005) (5) para diferenciar las formas completa y parcial de la enfermedad:
Forma completa
- Episodio agudo o recurrente que compromete el nervio óptico y la médula espinal, simultáneamente o en forma aislada.
- Serología positiva para el NMO-IgG.
- Mielitis total o parcial; neuritis óptica uni o bilateral.
- Ausencia de síntomas de compromiso encefálico del SNC, excepto del hipotálamo o tallo.
- Curso monofásico o recurrente.
- Compromiso medular de tres segmentos en la resonancia de médula espinal (T2), aunque puede ser normal.
- Resonancia cerebral normal (puede haber compromiso en hipotálamo o tallo).
Forma parcial
Compromiso de neuritis óptica o mielitis transversa. Serología positiva para NMO-IgG.
Las características histopatológicas de las lesiones incluyen infiltración por polimorfonucleares, desmielinización, pérdida de axones, eosinofilia, predominio de necrosis, inflamación e intensa respuesta perivascular (Figura 3). A nivel de la médula espinal se encuentra desmielinización, cavitación, destrucción y necrosis, en compromiso segmentario (4, 5, 10, 14).
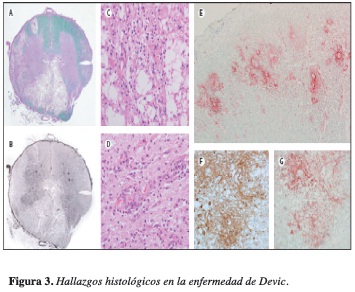
El autoanticuerpo NMO-IgG ha demostrado que el espectro de desórdenes de la NMO es más amplio de lo que previamente se creyó, e incluye algunos pacientes con episodio único o mielitis extensa longitudinal recurrente, neuritis óptica aislada recurrente, esclerosis múltiple ópticoespinal asiática y pacientes con enfermedad autoinmune coexistente tal como lupus eritematoso sistémico o síndrome de Sjögren (3,19).
El reciente descubrimiento de este autoanticuerpo NMO-IgG, altamente específico contra la barrera hematoencefálica, permitió redefinir los criterios diagnósticos y clasificar esta enfermedad como una canalpatía autoinmune (channelopathy) (20).
Las pruebas para detectar anticuerpos IgG contra aquaporina 4 (AQP4) tienen una sensibilidad de 73% y una especificidad de 91%. No obstante, estas pruebas aún no se comercializan en Colombia (sólo están disponibles por envío certificado a la Clínica Mayo en Estados Unidos, y son de muy alto costo).
El tratamiento busca evitar la cuadriparesia, la afectación del nervio óptico y la falla ventilatoria. Se utiliza metil-prednisolona 1 g diario por cinco días seguido de prednisolona oral. La plasmaféresis está indicada cuando no hay mejoría con los esteroides. También se ha intentado el uso de interferón Beta 1B y azatioprina en dosis de 2-3 mg/kg/día (8).
Los estudios inmunopatológicos sugieren que la pérdida immune de la acuaporina 4 es detectable en las lesiones tempranas de NMO. El anticuerpo monoclonal célula-B específico (CD20), rituximab, puede ser un tratamiento efectivo, aún en pacientes que no responden a otros fármacos (4, 5, 21).
En el presente, los corticoides son ampliamente utilizados como la primera línea de tratamiento de los ataques de neuritis óptica y mielitis, mientras que la plasmaféresis terapéutica se aplica en casos de falla a esteroides. Se han empleado otras estrategias para la prevención de recurrencias de NMO en pequeñas series de casos (mitoxantrone, micofenolato, inmunoglobulina) con resultados modestos (6, 12, 22).
El pronóstico visual ambulatorio de la NMO es reservado, por lo que debe asegurarse un tratamiento agresivo temprano para prevenir las recurrencias que resultan en severa discapacidad (13). Los predictores de mortalidad son: la asociación a enfermedades autoinmunes, episodios repetitivos en los dos primeros años de la enfermedad y persistencia de compromiso motor después del primer episodio (17).
Las características del cuadro clínico de esta paciente, con mielitis y neuritis óptica bilateral (que es infrecuente en EM), la resonancia magnética medular inicial que reportó lesión única cervical con compromiso de tres cuerpos vertebrales (cuyas imágenes no estuvieron disponibles en la actual hospitalización), la resonancia magnética cerebral sin criterios de EM, así como el curso clínico y evolución de la paciente, compatibles con NMO, permitieron establecer en este caso el diagnóstico de enfermedad de Devic, basado en los criterios de Wingerchuk, pese a no contar con la determinación de los autoanticuerpos NMO-IgG, por las circunstancias ya mencionadas. Desafortunadamente, el desenlace del compromiso visual en esta paciente fue catastrófico.
Conclusiones
- La NMO tiene características clínicas, inmunopatológicas e imaginológicas que permiten distinguirla de la EM.
- A diferencia de la neuritis óptica de la EM, en la NMO ésta puede ser fulminante y devastadora.
- La mielitis también puede ser fulminante, con retención urinaria aguda, paraparesia o cuadriparesia, severos espasmos tónicos e intenso dolor de espalda.
- La demostración de ausencia de bandas oligoclonales en LCR es útil para diferenciar NMO de EM.
- La serología para NMO-IgG por inmunofluorescencia indirecta es una herramienta valiosa para el diagnóstico de NMO, con sensibilidad de 73% y especificidad de 91%.
- El temprano reconocimiento de la forma recurrente de la NMO permite un tratamiento expedito agudo y crónico para prevenir la importante morbimortalidad de la enfermedad.
Agradecimientos
A la Unidad de Neurología del Hospital Universitario de La Samaritana, por su colaboración en la recolección de la información y análisis crítico de este trabajo.
Referencias
1. Jacob A, Bogglid M. Neuromyelitis optica. Practical Neurology 2006; 6: 180- 84. [ Links ]
2. Plant GT. Optic neuritis and multiple sclerosis. Curr Opin Neurol 2008; 21: 16-21. [ Links ]
3. Cross SA. Rethinking Neuromyelitis Optica (Devic disease). J Neuro-Ophthalmol 2007; 27: 57-60. [ Links ]
4. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neur 2007; 6: 805-15. [ Links ]
5. Mandler RN. Neuromyelitis optica - Devic's syndrome, update. Autoimmun Reviews 2006; 5: 537-43. [ Links ]
6. Daza J, Roncallo A. Neuromyelitis optica: state of the art. Salud Barranquilla 2007; 23: 204-19. [ Links ]
7. Weinshenker BG. Neuromyelitis optica is distinct from multiple sclerosis. Arch Neurol 2007; 64: 899-901. [ Links ]
8. Wingerchuk DM. Diagnosis and Treatment of Neuromyelitis Optica. The Neurologist 2007; 13: 2-11. [ Links ]
9. Galetta SL, Bennett J. Neuromyelitis optica is a variant of multiple sclerosis. Arch Neurol 2007; 64: 901-3. [ Links ]
10. Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG. Brain abnormalities in Neuromyelitis Optica. Arch Neurol 2006; 63: 390-6. [ Links ]
11. Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 2007; 68: 603-5. [ Links ]
12. Matiello M, Jacob A, Wingerchuk DM, Weinshenker BG. Neuromyelitis optica. Curr Opin Neurol 2007; 20: 255-60. [ Links ]
13. Behbehani R. Expanding the spectrum of neuromyelitis optica: firend or foe? Curr Opin Ophthalmol 2007; 18: 459-62. [ Links ]
14. Wingerchuk DM, Lucchinetti CF. Comparative immunopathogenesis of acute disseminated encephalomyelitis, neuromyelitis optica and multiple sclerosis. Curr Opin Neurol 2007; 20: 343-50. [ Links ]
15. Wingerchuk DM. Neuromyelitis optica: new findings on pathogenesis. Int Rev Neurobiol 2007. 79: 665-88. [ Links ]
16. Pittock SJ, Lennon VA, De Seze J, Vermersch P, Homburger HA, Wingerchuk DM et al. Neuromyelitis optica and Non-Organ-Specific Autoimmunity. Arch Neurol 2008; 65: 78-83. [ Links ]
17. Wingerchuk DM, Weinshenker BG. Neuromyelitis optica: Clinical predictors of a relapsing course and survival. Neurology 2003; 60: 848-53. [ Links ]
18. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66: 1485-9. [ Links ]
19. Weinshenker BG, Wingerchuk DM. Neuromyelitis optica: clinical syndrome and the NMO-IgG autoantibody marker. Curr Top Microbiol Immunol 2008; 318: 343-56. [ Links ]
20. Lalive PH, Perrin L, Chofflon M. Neuromyelitis optica/Devic's syndrome: new perspectives. Rev Med Suisse 2007; 3: 950-5. [ Links ]
21. Cree BAC, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005; 64: 1270-2. [ Links ]
22. Argyriou AA, Makris N. Neuromyelitis optica: a distinct demyelinating disease of the central nervous system. Acta Neurol Scand 2008; 118: 209-17. [ Links ]

