










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.35 no.2 Bogotá Apr./June 2010
ACTUALIZACIONES
Role of oxidative and nitrosative stress in cardiac hypertrophy and ventricular remodeling
Alejandro Giraldo(1) • Medellín
(1) MD, PhD. Director Biología de Sistemas y Laboratorio de Proteómica, Escuela Ciencias de la Salud, Universidad Pontificia Bolivariana, Medellín, Colombia.
Correspondencia. Director Biología de Sistemas y Laboratorio de Proteómica, Escuela Ciencias de la Salud, Universidad Pontificia Bolivariana. Calle 78B N° 72A 109, Medellín, Colombia. Tel. (+574) 4936300 Ext. 893.
E-mail: alejandro.giraldo@upb.edu.co - mdmbagiraldo@gmail.com
Recibido: 15/IX/09 Aceptado: 18/II/10
Resumen
Objetivo: hacer una revisión de los mecanismos moleculares del estrés oxidativo y nitrosativo en la fisiopatología de la falla cardiaca.
Metodología: se hizo una búsqueda en Medline (Pubmed) con las palabras clave: oxidative stress, ventricular remodeling, heart failure y nitrosative stress. Se consultó además bibliografía citada por autores de reconocida trayectoria en investigación en este tema.
Resultados: se seleccionaron los 112 artículos más relevantes en el tema de estrés oxidativo/ nitrosativo que se relacionarán con falla cardiaca.
Conclusiones: las respuestas de estrés de los cardiomiocitos y del tejido miocárdico es muy probable que constituyan un aspecto significativo del desarrollo de patologías cardiacas que desencadenan como evento final su progresión hacia falla cardiaca. El estrés oxidativo es un tema común en la fisiopatología de la cardiomiopatía isquémica y no isquémica. Patologías cardiacas como la falla cardiaca son usualmente precedidas de hipertrofia cardiaca secundaria, al menos en parte, a la generación de especies reactivas del oxígeno en los cardiomiocitos. Varios son los mecanismos implicados en la remodelación ventricular y progresión de la falla cardiaca que dependen de alteraciones homeostáticas en los sistemas que generan estrés oxidativo y/o nitrosativo. La discusión se centra en la reciente evidencia derivada de investigaciones llevadas a cabo tanto in vitro en cardiomiocitos cultivados como in vivo en modelos experimentales de patologías cardiacas. Las implicaciones clínicas de los recientes descubrimientos aunque son muy prometedoras para la terapéutica de la falla cardiaca, aun no han logrado trasladarse con total éxito a la práctica clínica en los ensayos clínicos realizados hasta el momento (Acta Med Colomb 2010; 2010; 35: 82-95).
Palabras clave: falla cardiaca, estrés oxidativo, remodelación ventricular, hipertrófica cardiaca, NAD(P)H oxidasas.
Abstract
Objective: to review the molecular mechanisms of oxidative and nitrosative stress in the process of ventricular remodeling and the pathophysiology of heart failure.
Metodology: a Medline (PubMed) search was performed using the keywords: oxidative stress, ventricular remodeling, heart failure, nitrosative stress. Cited bibliography in key papers from renowned authors in this field of research was also examined.
Results: 112 articles were selected as the most relevant regarding the several systems of oxidative stress production in the cardiovascular system in addition to their relevance to the pathophysiology of heart failure.
Conclusions: the stress responses of cardiomyocytes and the myocardial tissue as a whole are likely to constitute a significant aspect of the development of cardiac pathologies. Oxidative stress is a common theme in the pathophysiology of ischemic and non-ischemic cardiomyopathy. Cardiac pathologies such as heart failure are usually preceded by cardiac hypertrophy secondary, at least in part, to the generation of reactive oxygen species in cardiomyocytes. Several mechanisms have been implicated in ventricular remodeling during heart failure progression which depends on homeostatic alterations in oxidative and nitrosative stress. The discussion is centered in the exposition of recent evidence from research carried out either in vitro in cultured cardiomyocytes as well as from in vivo models of experimental cardiac pathologies. The clinical implications of this novel understanding, although very promising for the therapeutic treatment of heart failure, up until now, have failed to translate with total success to the clinical practice in several clinical trials (Acta Med Colomb 2010; 2010; 35: 82-95).
Keywords: heart failure, oxidative stress, ventricular remodeling, cardiac hypertrophy, NAD(P) H oxidases.
Introducción
Las enfermedades cardiovasculares ocupan el primer lugar de mortalidad en países desarrollados causando una de cada tres muertes (36%) (1, 2). Igualmente, en países en desarrollo como Colombia, las enfermedades cardiovasculares ocupan el primer renglón de morbimortalidad (3, 4). La falla cardiaca es un síndrome clínico de altísima prevalencia en el que confluyen la mayoría de las enfermedades que afectan en forma aguda o crónica al sistema cardiovascular. En Europa se ha estimado que mientras al menos 10 millones de pacientes tienen falla cardiaca sintomática, un número similar de pacientes presenta disfunción ventricular asintomática (5). Por su parte en los Estados Unidos hay al menos 5 millones de pacientes con diagnóstico de falla cardiaca y anualmente se diagnostican mas de 500 mil nuevos casos de esta enfermedad (6, 7). Todo ello hace que la falla cardiaca sea considerada un grave problema de salud pública que consume altos porcentajes de los recursos de salud de las naciones. El síndrome de falla cardiaca se caracteriza por pérdida progresiva de la función de bomba cardiaca asociado a síntomas como disnea progresiva, edema y fatigabilidad fácil. A pesar de los avances en el tratamiento farmacológico y los recientes desarrollos en el tratamiento no farmacológico de la falla cardiaca (p. ej. mallas antirremodelación, terapia de resincronización, dispositivos de asistencia ventricular) la mortalidad por esta enfermedad sigue siendo muy alta. Se estima que la expectativa de vida de los pacientes luego del diagnóstico de la enfermedad es muy reducido llevando a una mortalidad de casi el 50% a los cinco años. Estas cifras son aun más desalentadoras para los pacientes con formas severas de la enfermedad quienes llegan a tener una mortalidad de hasta el 50% luego del primer año de hecho el diagnóstico (5). Adicional a esto, el número de casos de falla cardiaca continúa en aumento progresivo como resultado de la mayor supervivencia de pacientes que han presentado infarto de miocardio (IAM) y la tendencia global a una mayor expectativa de vida, con un incremento consecuente en la población anciana en la que la prevalencia de falla cardiaca es muy alta (2).
El hecho de que la polifarmacia logre mejorar un poco las expectativas de vida de los pacientes con falla cardiaca pero con un impacto mínimo respecto a lo que sería deseable, indica que la falla cardiaca sigue siendo progresiva a pesar de un tratamiento farmacológico óptimo. Esto sugiere que aun no se ha logrado un entendimiento global de la falla cardiaca y que por tanto existen blancos terapéuticos claves que están por ser descubiertos y cuya intervención podría lograr modificaciones dramáticas del curso de la enfermedad.
Evidencia de que la falla cardiaca es en principio reversible, lo constituyen los varios informes en la literatura que indican la casi total reversión de la falla cardiaca observada en pacientes que se han sometido al uso de dispositivos de asistencia ventricular por un tiempo prolongado (8). Aunque estos resultados dan esperanzas a los pacientes con falla cardiaca, los altos costos, su falta de disponibilidad universal, y las complicaciones y riesgos derivados de su uso, impiden su aplicación como terapia en la mayoría de los pacientes (8). Por tanto, existe una urgente necesidad de ganar un mejor entendimiento de los mecanismos fisiopatológicos moleculares de la falla cardiaca que ayuden al desarrollo de estrategias farmacológicas novedosas orientadas a detener la progresión de esta devastadora enfermedad. Recientemente se ha avanzado sustancialmente en el entendimiento de los mecanismos moleculares que regulan la producción de especies reactivas de oxígeno (ROS, de la sigla en inglés reactive oxygen species) en el sistema cardiovascular, al igual que de los efectos que el mal funcionamiento de estos mecanismos tiene en la fisiopatología de la hipertrofia cardiaca y la remodelación ventricular y que finalmente lleva a falla cardiaca. Esta revisión hará una síntesis de los diferentes mecanismos de producción de ROS en el sistema cardiovascular implicados en estos procesos recientemente dilucidados. Sin embargo, para mayor claridad de esta exposición, inicialmente se hará una breve discusión del paradigma actual del entendimiento de la fisiopatología de la hipertrofia cardiaca y su progresión hacia la falla cardiaca.
Hipertrofia cardiaca
En el sentido más general, la hipertrofia cardiaca es una respuesta adaptativa a demandas hemodinámicas incrementadas. Los cardiomiocitos (las células contráctiles del corazón) se encuentran terminalmente diferenciados y por tanto no entran al ciclo celular ni tienen citoquinesis luego del nacimiento (9, 10). Es por ello que la hipertrofia cardiaca es el resultado del crecimiento celular de cardiomiocitos (p. ej. incremento en el contenido celular de miofibrillas) en ausencia de división celular. No obstante, investigaciones recientes han mostrado que existen células cardioprogenitoras residentes en el tejido miocárdico y también provenientes de la sangre periférica las cuales pueden regenerar el tejido miocárdico (11, 12). Pese a ello, en términos generales, la capacidad regenerativa de estas células cardioprogenitoras, por ejemplo luego de un IAM, es mínima y por consiguiente la mayor cantidad de tejido asociado a una cicatriz de un IAM lo constituye tejido fibrótico con pobres propiedades contráctiles.
Los genes inmediatos tempranos son una clase de genes cuya expresión es aguda y usualmente transitoria, precedida de activación de cascadas de señalización intracelular y ocurre en ausencia de síntesis proteica de novo (13). En cardiomiocitos, el fenotipo hipertrófico se encuentra asociado a cambios en la expresión de genes inmediatos tempranos (p. ej. c-jun, ATF3, IL-6) como lo hemos demostrado recientemente (14) y la reexpresión de un patrón de genes que usualmente se expresa durante el desarrollo embrionario del corazón. Este último es el llamado "patrón fetal" de expresión de genes cardiacos, que incluye al péptido natriurético auricular, péptido natriurético tipo B, la cadena pesada de miosina tipo β, entre otros, y que son usados frecuentemente como marcadores de estrés cardiaco y de hipertrofia cardiaca en modelos experimentales (15, 16).
El patrón de la hipertrofia cardiaca es dependiente del estímulo
Dos tipos diferentes de sobrecarga hemodinámica llevan a dos patrones bien diferenciados de hipertrofia cardiaca. Así, la sobrecarga de presión, como ocurre en los estados hipertensivos, lleva al desarrollo de hipertrofia cardiaca concéntrica en la que el espesor de la pared ventricular se incrementa, sin modificaciones o modificaciones mínimas del diámetro de la cámara ventricular (17). Debido a la disposición circunferencial de los cardiomiocitos en las fibras musculares cardiacas, este patrón de hipertrofia se correlaciona con un incremento en el área de sección transversal de los cardiomiocitos, como resultado de una disposición de las sarcómeras en paralelo (17). En contraste, la sobrecarga de volumen, como ocurre en la insuficiencia mitral o aórtica, lleva al desarrollo de hipertrofia excéntrica que se caracteriza por un incremento en el diámetro de la cámara ventricular acompañado por un incremento proporcional del espesor de la pared. A nivel celular, este patrón de hipertrofia se correlaciona con un incremento proporcional en el área de sección transversal y de la longitud del cardiomiocito, y una disposición de sarcómeras tanto en serie como en paralelo. Estos dos patrones de hipertrofia cardiaca son necesarios para lograr un estado compensado respecto a la sobrecarga hemodinámica impuesta, manteniendo de esta forma el estrés sistólico de la pared ventricular en límites normales de acuerdo con la ley de Laplace (17).
La distinción entre hipertrofia concéntrica y excéntrica no es trivial por varias razones. La adaptación específica de la estructura del ventrículo a una carga hemodinámica particular virtualmente prohíbe la aparición de condiciones aberrantes como el desarrollo de hipertrofia excéntrica y el incremento correspondiente en la longitud del cardiomiocito en respuesta a sobrecarga de presión. Tal respuesta sería claramente maladaptativa al no compensar hemodinámicamente las demandas impuestas sobre el ventrículo, debido a un aumento desmesurado de la presión de la pared del ventrículo de acuerdo con la ley de Laplace (17). A este respecto es importante anotar que datos cumulativos recientes indican que alteraciones intrínsecas en los cardiomiocitos, que incluye elongación de los cardiomiocitos, y que son colectivamente conocidas como remodelación de los cardiomiocitos (18), contribuyen al proceso conocido como remodelación ventricular y a la progresión de la falla cardiaca (ver más adelante) (19, 20).
Fisiopatología de la falla cardiaca
Una revisión completa de la fisiopatología y bases moleculares de la falla cardiaca está más allá de los alcances de esta corta revisión. A continuación se enunciarán los conceptos que explican bajo el paradigma actual de conocimientos fisiopatológicos y moleculares la aparición de la falla cardiaca como síndrome clínico crónico. El paradigma de la fisiopatología de la progresión de la falla cardiaca fue propuesto hace más de 40 años por Felix Meerson (20, 21). Así, luego de un insulto inicial hacia el corazón (Estadio 1 o "avería transitoria"), bien sea por incremento en las demandas hemodinámicas como en la hipertensión arterial, o pérdida de tejido como sucede en el IAM, el corazón se hipertrofia de manera compensadora para mantener el gasto cardiaco dentro de límites normales (estadio 2 o "hiperfunción compensatoria"). Sin embargo, cuando el estímulo inicial persiste como sucede en la hipertensión no controlada o es irreversible como en el IAM, una fase final denominada "fatiga crónica del miocardio" (estadio 3) tiene lugar durante meses a años (20-22). Este estadio 3 se caracteriza por falla ventricular progresiva (con reducción paulatina de la fracción de eyección), incremento de la hipertrofia de cardiomiocitos (principalmente a expensas de elongación que lleva a dilatación cardiaca) y reemplazo de cardiomiocitos muertos (debido a necrosis o apoptosis) por tejido fibrótico (22). El desenlace clínico de este proceso es descompensación cardiaca progresiva y falla cardiaca manifiesta. A partir de los estudios de Meerson queda claro que, luego de un estadio de hipertrofia compensatoria, con un periodo clínicamente estable que dura entre varios meses a varios años, el incremento crónico de las demandas hemodinámicas sobre el corazón, invariablemente llevará a una fase de "fatiga crónica" que finalmente se manifestará como el síndrome clínico de falla cardiaca.
Remodelación ventricular: el camino hacia la falla cardiaca
La fisiopatología molecular de la falla cardiaca ha probado ser multifactorial y compleja. Los mecanismos moleculares íntimos de la progresión de la falla cardiaca han estado bajo intensa labor de investigación en las pasadas dos décadas. El peso de la evidencia acumulada apoya el concepto de que este proceso es el resultado final de la estimulación neurohumoral persistentemente incrementada, la cual activa múltiples cascadas de señalización intracelular que cambian el fenotipo del miocardio al alterar los patrones de expresión de genes que controlan el crecimiento (hipertrofia) y el balance entre apoptosis y supervivencia de los cardiomiocitos (13, 14). El fenotipo resultante se caracteriza por pobre contractilidad, alteraciones energéticas miocárdicas, fibrosis miocárdica, y dilatación ventricular con un incremento correspondiente en la longitud del cardiomiocito (elongación) (23-25). A este proceso que lleva a falla cardiaca progresiva se le ha denominado remodelación ventricular y fue establecido como componente paradigmático de la fisiopatología de la falla cardiaca por los estudios pioneros de Janice Pfeffer hacer mas de 30 años, usando modelos animales de sobrecarga hemodinámica (p. ej. ratas espontáneamente hipertensas), y de isquemia miocárdica inducida por ligación de arterias coronarias (26-29).
Estrés oxidativo y falla cardiaca
Los cardiomiocitos, las células contráctiles del corazón, poseen una capacidad muy limitada para regenerarse y es por ello que el corazón es particularmente vulnerable al daño inducido por diferentes agentes nocivos, al igual que eventos secundarios a isquemia o isquemia acoplada con reperfusión. Los cardiomiocitos pueden morir por necrosis y/o apoptosis secundaria a la producción incrementada de ROS. La producción de ROS en el corazón ocurre durante la isquemia (30, 31), pero se incrementa aún más luego de reperfusión (30, 32). No obstante, aun en ausencia de enfermedad coronaria o cardiopatía isquémica, la masa incrementada del ventrículo durante la hipertrofia cardiaca (o alteraciones intrínsecas durante la diástole en el modelo de falla cardiaca inducido por taquicardia con marcapasos) hacen que el corazón sea vulnerable de sufrir episodios de isquemia debido a una reducción en la reserva coronaria. Esto último es particularmente importante en el subendocardio debido al incremento del estrés de la pared durante la hipertrofia ventricular (33-37). La producción de ROS también se ha implicado en varios procesos subagudos en los que la producción de ROS en la mitocondria y fuera de la mitocondria por enzimas citoplasmáticas como la NADPH oxidasa o la xantina oxidasa parecen estar involucradas en el desarrollo de hipertrofia, remodelación ventricular y progresión de la falla cardiaca.
Fuentes de ROS y efectos sobre hipertrofia y remodelación cardiaca
Varias fuentes de ROS se han identificado en el corazón, incluyendo la mitocondria, las xantina oxidasas, óxido nítrico sintetasas tipo 3 (ONS3) desacopladas, las NADPH oxidasas (NOX), y células infiltrativas inflamatorias. Es de resaltar que las enzimas NOX parecen tener un papel primordial en la señalización redox del corazón y se han implicado en procesos como hipertrofia en respuesta a sobrecarga de presión y remodelación ventricular luego de IAM.
La mitocondria como fuente de ROS en el sistema cardiovascular
Bajo condiciones normales, más del 1%-2% de la captación de O2 por la mitocondria lleva a la producción de ROS (38,39). Esta tasa suele incrementarse durante la isquemia y aun más durante la isquemia acoplada con reperfusión (30-32). Estas condiciones tienen alto impacto en el corazón dado que hasta el 40% del volumen de los cardiomiocitos es ocupado por mitocondrias (40, 41). El átomo de oxígeno contiene dos electrones no apareados en su orbita externa, y es por tanto altamente reactivo. Esto hace que el O2 tienda a iniciar reacciones en cadena que resultan en la producción de varios tipos de ROS los cuales pueden inducir cambios químicos irreversibles en lípidos y proteínas celulares y por tanto daño celular.
En el metabolismo normal, radicales de anión superoxido (O2-) se producen durante la respiración celular como un subproducto de la reducción monoelectrónica del O2 (42-44). La reducción completa de O2 a H2O, el evento final en la cadena de transporte de electrones en la mitocondria, requiere la donación de otros tres electrones una vez formado el O2- (Figura 1). Así, O2-es rápidamente metabolizado en la mitocondria por la enzima manganeso superóxido dismutasa (Mn2+-SOD) y en el citosol por la Cu2+/Zn2+-SOD, para formar entonces peróxido de hidrógeno (H2O2), un agente oxidante menos potente pero más estable (45) (Figura 1). El H2O2 es usualmente removido por la acción de la catalasa, glutatión peroxidada (GPx) o la peroxirredoxina (46-48). Sin embargo, en condiciones patológicas, en presencia de iones de cobre o hierro, H2O2 forma el radical hidroxilo (·OH) que es altamente reactivo (Figura 1), en un proceso que puede estar facilitado por la liberación de hierro ferroso a partir de enzimas como la aconitasa, que contienen el núcleo [Fe4S4]2+ cuando esta enzima es dañada por el O2- (49) (Figura 1).
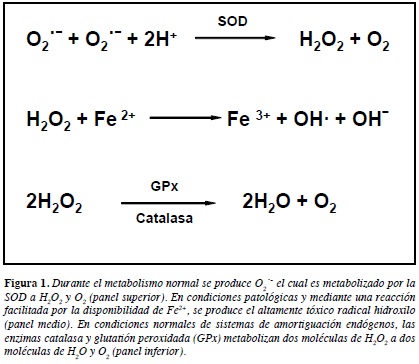
La evidencia del importante papel de la mitocondria en la producción de ROS se deriva de experimentos en los que se ha observado el eflujo de H2O2 en mitocondrias intactas así como también en órganos prefundidos, sugiriendo que la mitocondria produce O2- in vivo, el cual es luego reducido a H2O2 (50). La producción de O2- por la mitocondria se ha atribuido a los complejos mitocondriales I y III (43, 51, 52).
La producción incrementada de ROS se ha documentado a partir de cardiomiocitos derivados de modelos animales de falla cardiaca inducida por marcapasos en perros y en falla cardiaca inducida por IAM en ratones (51, 53, 54). Las mitocondrias son también una fuente de ROS (a partir del complejo mitocondrial III) en cardiomiocitos expuestos a factor de necrosis tumoral α (TNFα) y esto lleva a disfunción mitocondrial y daño del DNA mitocondrial. Esto sugiere que uno de los efectos patológicos del TNFα durante la remodelación ventricular se deriva, al menos en parte, de la inducción de la producción de ROS y disfunción mitocondrial secundaria (55). Es importante destacar que H2O2 se ha documentado como la molécula de señalización responsable de la muerte de células cultivadas luego de su exposición a TNFα (56).
Bajo condiciones fisiológicas normales la producción de ROS por la mitocondria es relativamente baja (57), sin embargo, se ha descrito un fenómeno de liberación de ROS inducida por ROS (LRIR) en la mitocondria, en el cual la mitocondria responde a concentraciones aumentadas de ROS (sean éstas endógenas o exógenas), incrementando su propia producción de ROS (58-60). En la mitocondria, al menos dos mecanismos diferentes se han involucrado en la LRIR, uno de ellos involucra a la activación del poro de transición de la permeabilidad mitocondrial (PTPM), y el segundo mecanismo involucra la apertura del canal aniónico mitocondrial interno (CAMI) (61). El PTPM es un poro mitocondrial inespecífico que atraviesa ambas membranas de la mitocondria y que contiene el canal aniónico voltaje dependiente (CAVD, también conocido como porina) en la membrana externa, la translocasa de nucleótidos de adenina (TNA) en la membrana interna, y ciclofilina D en la matriz, probablemente en asocio con el receptor de benzodiacepina, el cual parece estar en contacto estrecho con el CAVD en la membrana externa (62-64). La LRIR a través de PTPM lleva a un aumento súbito de la producción de ROS desde la cadena de transporte de electrones, un mecanismo que puede estar involucrado en la regulación de las vías apoptóticas y de precondicionamiento en los cardiomiocitos (65, 66). En el segundo mecanismo de LRIR, la apertura del CAMI secundaria a elevadas concentraciones de ROS también resulta en el súbito y transitorio incremento de ROS derivados de la cadena de transporte de electrones. Este último mecanismo puede estar involucrado en el desencadenamiento de arritmias cardiacas luego de isquemia miocárdica (67).
La mayoría de las especies de ROS como O2- y ·OH poseen capacidades limitadas para la difusión, probablemente debido a su corta vida media. Es por ello que sus acciones son limitadas a un corto periodo de tiempo y confinadas en el espacio. Por el contrario, H2O2 es una forma difusible de ROS que es a su vez más estable. Es de anotar que H2O2 puede ser el mediador clave de la propagación de la señal de apoptosis en cardiomiocitos a través de la amplificación de la respuesta en ondas de activación entre una y otra mitocondria por medio de la LRIR mediada por PTPM (68, 69). Igualmente como se mencionó anteriormente, probablemente por su mayor y más rápida difusión, H2O2 se ha postulado como la molécula de señalización responsable de la apoptosis en células cultivadas expuestas a TNFα (56).
En experimentos in vitro, la exposición de cardiomiocitos a concentraciones crecientes de H2O2 tiene diferentes efectos en los cardiomiocitos. Así, altas concentraciones de H2O2 (>0.5 mM) causan muerte celular necrótica (70), mientras que concentraciones intermedias (0.1 - 0.5 mM) promueven un fenotipo apoptótico (71,72). Por el contrario, concentraciones bajas de H2O2 (≤ 0.04 mM) no son tóxicas, y por el contrario promueven citoprotección y crecimiento hipertrófico de los cardiomiocitos (70). Es por ello que el H2O2 constituye un ejemplo relevante de estrés oxidativo fácilmente manipulable en ambientes de laboratorio experimental.
Producción de ROS por las xantina oxidasas
La enzima xantina oxidorreductasa (XOR) es codificada por un solo gen, pero la proteína existe como un homodímero en dos formas interconvertibles, xantina deshidrogenasa (XDH) y xantina oxidasa (XO) (73). Estas dos formas difieren en que XO reduce sólo oxígeno, mientras que XDH puede reducir al NAD+ además del oxígeno (74). Ambas formas de XOR son reducidas durante el proceso de síntesis de ácido úrico a partir de xantina durante el metabolismo de las purinas. El proceso de reoxidación de XOR es llevado a cabo en cuatro pasos secuenciales. Durante los primeros dos pasos, dos electrones son transferidos a O2, generando de esta forma H2O2 (75). En los dos pasos finales, los dos electrones restantes son transferidos separadamente a moléculas de O2 para generar O2-. Por tanto, cada ciclo de esta enzima genera dos moléculas de H2O2 y dos moléculas de O2- (76). Evidencia reciente sugiere que XOR se expresa a bajos niveles en cardiomiocitos (77), probablemente localizada en el retículo sarcoplásmico (78). La elevada expresión de XOR y un incremento en su actividad se ha documentado en falla cardiaca en humanos en fase terminal y en un modelo de falla cardiaca inducida por taquicardia con marcapaso (77, 79). Es de anotar que el tratamiento con el inhibidor de XO, alopurinol, mejora la contractilidad y disminuye el consumo miocárdico de oxígeno en el modelo de falla cardiaca inducido por taquicardia con marcapaso en perros (79, 80).
También hay evidencia que el alopurinol reduce significativamente la remodelación ventricular secundaria a IAM experimental en ratones (81), dando soporte adicional al papel de la XO en las patologías cardiacas. En un modelo de hipertrofia inducida por sobrecarga de presión en ratas, la actividad incrementada de XO sólo fue observada cuando la falla cardiaca se hizo manifiesta, mas no durante la hipertrofia compensada (82). Esto sugiere que en remodelación ventricular secundaria a sobrecarga de presión, la actividad de XO es más importante en los estadios avanzados y puede entonces realizar un papel importante en la transición hacia falla cardiaca (82). Recientemente, en un modelo de ratas espontáneamente hipertensas con falla cardiaca establecida, el tratamiento con oxipurinol por cuatro semanas disminuyó la actividad XO y la producción de superóxido, todo lo cual redundó en efectos benéficos evidenciados por reversión de la remodelación miocárdica del ventrículo izquierdo (83). A pesar de estos prometedores resultados en animales de experimentación, y estudios pilotos iniciales con un reducido número de pacientes con falla cardiaca (84-86), los estudios en humanos han fallado en trasladar los potenciales efectos benéficos de la terapia con inhibidores de la XO en el tratamiento de la falla cardiaca. De esta forma el estudio OPT-CHF publicado recientemente, y en el que se evaluó los efectos en un número importante de pacientes (n= 405), no mostró resultados benéficos en la mortalidad y pronóstico de pacientes con falla cardiaca moderada a severa (clase funcional NYHA III-IV) (87). Este estudio sólo mostró una mínima tendencia a la mejoría del pronóstico en pacientes que presentan niveles elevados de ácido úrico al inicio de la terapia (87).
NAD(P)H oxidasas y estrés oxidativo en el sistema cardiovascular
Las enzimas asociadas a membranas tipo NAD(P)H oxidasas (NOX), son una fuente importante de ROS en el sistema cardiovascular. Cada enzima contiene una subunidad catalítica denominada Nox, de las cuales hay 5 isoformas descritas (Nox 1-5), que son codificadas por genes separados (88). Cada subunidad Nox forma un heterodímero con la subunidad p22phox, constituyendo un núcleo catalítico heterodimérico (o citocromo b558) donde la transferencia de electrones a partir de NAD(P)H al O2 resulta en la formación de O2-. Cabe resaltar, que mientras Nox1 y Nox2 claramente requieren la asociación de subunidades reguladoras citosólicas (p47phox, p67phox, p40phox and Rac1) para activar la producción de O-, Nox4 no requiere de éstas (89, 90). La distribución tisular de las diferentes isoformas Nox en el sistema cardiovascular varía considerablemente. Así, Nox1 se expresa más abundantemente en las células de músculo liso vascular (91), por su parte Nox2 y Nox4 se encuentran abundantemente en cardiomiocitos (92, 93) y células endoteliales (94). En los fibroblastos se ha descrito la expresión de Nox2, Nox4 y Nox5 (95, 96).
Aunque durante el desarrollo de los cardiomiocitos Nox4 es la isoforma predominante con un papel en la diferenciación cardiaca (97), en el corazón adulto la isoforma predominante es Nox2. Nox2 está implicada en la regulación de vías de señalización redox-sensibles que modulan la actividad de kinasas y fosfatasas (98), la expresión de genes (99), y la inactivación de óxido nítrico (ON) dependiente de superóxido inducida por Ang II (100). Evidencia cumulativa indica que las enzimas NOX desempeñan un papel importante en la hipertrofia cardiaca, remodelación ventricular y falla cardiaca. En hipertrofia cardiaca inducida por sobrecarga de presión, la expresión y actividad de Nox2 se encuentra incrementada en cardiomiocitos y células endoteliales (92). En humanos la expresión de Nox2 se encuentra incrementada en cardiomiocitos luego del IAM (101), y en pacientes con falla cardiaca terminal (102).
Recientemente, ratones con knockout del gen Nox2 (Nox2 -/-) han aportado información importante respecto del papel de Nox2 en la hipertrofia y remodelación cardiaca. Los animales Nox2 -/- exhiben una respuesta hipertrófica atenuada en respuesta a dosis bajas de Ang II que no afectan la presión arterial (subpresoras) suministrada en infusión continua por un periodo de 7 a 14 días. Esta respuesta es paralela a una reducida actividad NOX, así como tambien una reducción en la expresión de ANP y disminución de la fibrosis miocárdica respecto a animales con el gen intacto (103) (Figura 2). A pesar del importante papel de Nox2 en la hipertrofia inducida por agonista, la situación es diferente durante la sobrecarga de presión. Allí, Nox2 puede realizar un papel preponderante en mediar la remodelación patológica. Así, ratones Nox2 -/- que fueron sometidos a constricción de la aorta torácica (CAT) desarrollaron hipertrofia cardiaca y expresión incrementada de ANP, la cual se correlacionó con un incremento paradójico de la actividad NOX, atribuida a Nox4 dada la ausencia de Nox2 (104). Sin embargo, la función sistólica se encontró preservada en los ratones Nox2 -/- sometidos a CAT, asociado a reducción en la fibrosis miocárdica respecto a ratones sin modificaciones genéticas (104). Más recientemente, el mismo grupo reportó un análisis más detallado de la función ventricular contráctil en ratones Nox2 -/- comparados con ratones genéticamente intactos luego de CAT. En este estudio se demostró que los animales Nox2 -/- se encuentran protegidos de la disfunción sistólica y diastólica asociada a CAT tanto en el corazón como en los cardiomiocitos aislados (105) (Figura 2).
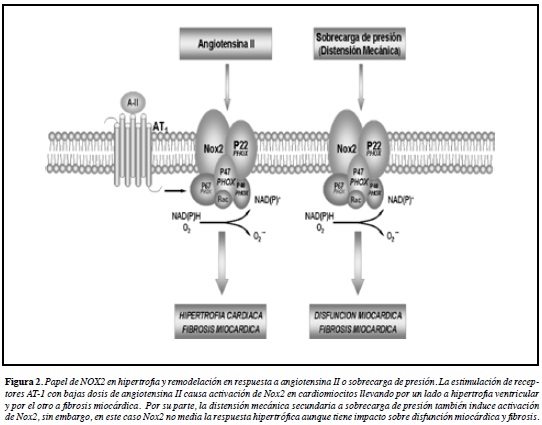
Es importante destacar que la actividad NOX es significativamente incrementada en respuesta a varios estímulos relevantes para la fisiopatología de la hipertrofia cardiaca, remodelación ventricular y falla cardiaca. Estos estímulos incluyen distensión mecánica (106), Ang-II (107), ET-1 (108), agonistas α-adrenérgicos (109) y TNF-α (55). Así mismo, se ha descrito que la translocación de la subunidad p47phox es importante para la activación de la enzima Nox2 en respuesta a Ang II (110). Consistente con esto, ratones knockout p47phox -/- en quienes se indujo IAM mediante ligación de coronarias exhibieron reducción en la dilatación ventricular con reducción significativa en la mortalidad. Este efecto estuvo asociado con reducción en la apoptosis, disminución de la actividad gelatinolítica de MMP2 y preservación de la producción de ON endotelial, al comparar con animales genéticamente intactos que también fueron sometidos a IAM (111). Cabe destacar que no sólo la actividad NOX sino además la actividad XO se vieron reducidas en ratones p47phox -/- luego de IAM comparado con animales sin esta modificación genética (111), confirmando estudios previos in vitro que sugieren que la actividad XO probablemente es iniciada secundaria a la activación de NOX (112).
Desbalance nitroso/redox y producción de ROS en el corazón
Recientemente el balance nitroso-redox y sus alteraciones en el sistema cardiovascular han recibido considerable atención debido a que pueden explicar varios aspectos de la fisiopatología de la falla cardiaca (113). Así, se ha sugerido que el desacoplamiento de la ON sintetasa 3 (ONS3, también conocida como ONS endotelial) es una importante fuente de ROS en el sistema cardiovascular, y esto podría ser también responsable de la disfunción endotelial que es un hallazgo frecuente en los pacientes con falla cardiaca (114). En el sistema cardiovascular normal las concentraciones de ROS y las especies reactivas del nitrógeno (ERN) son mantenidas dentro de los límites normales, permitiendo la óptima señalización dentro de la célula (115). Un incremento en ROS o una disminución en ERN alterará entonces esta señalización y desencadenará un desbalance nitroso redox con efectos deletéreos para el sistema cardiovascular (116). El que la ONS3, la isoforma predominante de ONS en cardiomiocitos y células endoteliales (117), constituya una fuente importante de ROS es intrigante dado que la cascada de eventos activada por el ON, como lo es la activación de la síntesis de guanosina monofosfato cíclico (cGMP) por la guanilato ciclasa y la consecuente activación de la proteína kinasa G (PKG), están asociadas a disminución de la hipertrofia cardiaca y de la remodelación ventricular (118,119). Los mecanismos moleculares involucrados en estas acciones incluyen modulación de la expresión de genes, estado de fosforilación de proteínas blanco y supresión de eventos de señalización activados por agonistas de receptores acoplados a proteínas Gq/11 (120-122). Sin embargo, O2- al reaccionar con ON forma peroxinitrito (ONOO-), un poderoso oxidante, desencadenando en esta forma estrés nitrosativo y una reducción en la disponibilidad y bioactividad del ON, disminuyendo en esta forma sus efectos antihipertróficos y antirremodelación (123). Bajo estas condiciones, ONS3 se convierte además en un generador de O2- en lugar de un generador de ON, agravando en esta forma el desbalance nitroso-redox (124). Evidencia reciente indica que la generación de O2- por la ONS es un evento fisiológico que ocurre normalmente y que se mantiene bajo control por la tetrahidrobiopterina (BH4). Éste además puede ser un paso intermedio en la síntesis de ON por la ONS (125).
Bajo condiciones fisiológicas normales, NOS3 usa NAD(P)H para generar ON y L-citrulina a partir de L-arginina y O2. Los electrones son transferidos desde el dominio reductasa al núcleo catalítico, y BH4 es el cofactor esencial para la donación de un electrón y un protón en una reacción cíclica (126). Calmodulina controla la translocación de electrones, mientras que un complejo zinc-tiolato así como la BH4 se requieren para la formación de un heterodímero de ONS3, el cual es esencial para la estabilidad del dominio oxidasa (estado acoplado) (127,128). Bajo condiciones de estrés oxidativo o nitrosativo incrementado o cuando se encuentra deprivada de su cofactor BH4 o su sustrato Larginina, la ONS3 se desacopla a una forma monomérica y en esta forma los electrones son desviados hacia O2 en lugar de L-arginina con un incremento subsecuente en la producción de O2- (129,130). El desacoplamiento de la ONS3 se ha reportado como un proceso patológico en el sistema vascular que parece estar involucrado en la fisiopatología de la disfunción endotelial en hipertensión y ateroesclerosis (131, 132).
El papel del desacoplamiento de la ONS3 en la remodelación cardiaca fue evaluado recientemente luego de sobrecarga de presión. Así, la CAT en ratones resultó en remodelación ventricular excéntrica patológica y disfunción ventricular asociada a disminución del homodímero de ONS3 en el miocardio e incremento en el estrés oxidativo (133). Consistente con un papel de la ONS3 en el remodelamiento patológico observado en estos animales, ratones deficientes en el gen ONS3 (ONS3-/-) luego de una CAT desarrollaron hipertrofia compensada más modesta, con menor dilatación ventricular y fibrosis intersticial asociado a menor estrés oxidativo (133). La generación de ROS por la ONS3 fue reducida en un 50% al preincubar extractos miocárdicos con el inhibidor NG-monometil-L-arginina (L-NMMA), sugiriendo que ROS fueron generados por la ONS3. Al comparar los ratones ONS3 -/- con los animales genéticamente intactos, estos últimos presentaron incremento marcado en la actividad gelatinasa de las MMP2 y MMP9 luego de la sobrecarga de presión (133), consistente con la observación que MMP2 y MMP9 son activadas por ROS en el corazón y están involucradas en la dilatación ventricular y progresión de la falla cardiaca secundario a la degradación de la matriz extracelular (134-138). El papel de la depleción de BH4 en el desacoplamiento de ONS3 fue también evaludo, así, la suplementación de BH4 a ratones sometidos a CAT resultó en hipertrofia concéntrica compensada similar a la observada en ratones deficientes en ONS3 también sometidos a CAT (133), sugiriendo que BH4 puede ser una atractiva medida terapéutica en falla cardiaca. Esto ha sido comprobado más recientemente en ratones que desarrollaron hipertrofia cuatro semanas después de inducir sobrecarga de presión mediante CAT. En los ratones que fueron suplementados con BH4 hubo reversión de la hipertrofia ventricular izquierda (HVI) y de la fibrosis miocárdica, mejoría de la disfunción ventricular y de cardiomiocitos, así como también reacoplamiento de la ONS3 y reducción del estrés oxidativo (139). Una observación interesante derivada de este estudio es que a pesar de los efectos benéficos derivados de la suplementación con BH4, la administración de BH4 desde el momento de iniciarse el estímulo hemodinámico no previno la aparición de hipertrofia durante la primera semana, aunque sí se previno la remodelación ventricular subsecuente (139). Cabe anotar que el desacoplamiento de ONS3 puede ocurrir incluso sin depleción de BH4 (140), y la consecuente producción de O2- puede entonces oxidar BH4, llevando a desacoplamiento de ONS3, formando así un proceso que se autoamplifica y que puede llevar a estrés oxidativo y/o nitrosativo y finalmente remodelación patológica cardiaca (141).
En otro estudio se indujo sobrecarga de presión en ratones mediante constricción de la aorta abdominal (CAA), resultando en HVI, dilatación y disfunción ventricular, la cual se correlacionó con expresión y actividad incrementada de MMP2 y MMP9, al comparar con animales operados sin inducción de CAA (138). Adicionalmente, la inducción de sobrecarga de presión por CAA en ratones que presentaban mutantes de colágeno que les concedía resistencia a las colagenasas, resultó en hipertrofia ventricular mínima, con reducción de la disfunción y el estrés de pared comparado con animales sin alteración genética que recibieron CAA (138). En este mismo estudio, el desbalance nistroso/redox fue sugerido por la reducida expresión de antioxidantes como tioredoxina y SOD (ver siguiente sección) y el incremento concomitante en la ONS inducible (ONSi, o ONS2), asociado a disminución de la ONS3 y a un incremento en proteínas nitrosiladas (138).
A pesar de que estos estudios muestran evidencia contundente del papel del desacoplamiento de ONS3 en la remodelación ventricular patológica en respuesta a sobrecarga de presión, otros estudios han mostrado resultados bastante contradictorios respecto a los ya presentados. Así, la ligación de arterias coronarias en ratones deficientes en ONS3 (ONS3 -/-) indujo una mayor disfunción, dilatación e hipertrofia ventricular comparado con animales sin alteración genética (142). Similarmente, la CAA (143) o CAT (144), resultó en mayor disfunción del ventrículo izquierdo en ratones deficientes en ONS3 comparado con animales genéticamente intactos sometidos al mismo estímulo hemodinámico. Consistente con esto, la restauración transgénica de la actividad ONS3 en los cardiomiocitos de ratones NOS3 -/- (ratones NOS3 -/-TG), resultó en atenuación de la HVI y disfunción ventricular luego de la CAT comparando con animales NOS3 -/- (145). Sin embargo, a pesar de no haber encontrado diferencias en la relación homodímeros/ monómeros de ONS3, la producción de ROS se encontraba incrementada en los animales NOS3 -/-TG luego de CAT (145). Las razones de estos resultados divergentes siguen siendo desconocidas, aunque pueden reflejar diferencias en la naturaleza y/o intensidad del estímulo de sobrecarga hemodinámica, y los diferentes escenarios que esto puede crear en términos de ONS3 acoplada vs desacoplada y la cantidad de ROS así producida.
Mecanismos antioxidantes
Dadas las acciones potencialmente deletéreas e inespecíficas que incrementos en la producción de ROS puede tener dentro de las células, un aspecto importante de la producción de ROS es el control estrecho que bajo condiciones fisiológicas, se ejerce sobre los sistemas responsables de su producción y los sistemas de amortiguamiento para remover ROS. Hay varios mecanismos celulares que regulan los niveles de ROS. Aquí discutiremos dos elementos de control claves en la regulación de ROS: SOD y el sistema Tioredoxina (Trx).
En el corazón, SOD se expresa en dos isoformas: Mn2+- SOD en la matriz mitocondrial, y Cu2+/Zn2+-SOD, en el citosol (146). La rápida producción de H2O2 y O2 a partir de O2- mediante SOD está usualmente acoplada con las acciones de las catalasas (en los peroxisomas tisulares), glutatión-peroxidasas (en el citoplasma) y las peroxiredoxinas (46-48,147) (Figura 1). Consistente con ello, ratones con sobrexpresión cardioespecífica de glutatión peroxidasa exhiben remodelación ventricular disminuida luego del IAM (148). Igualmente, la sobrexpresión cardioespecífica de Mn2+-SOD en ratones protege de daño miocárdico luego de isquemia/reperfusión (149), mientras que la recuperación funcional posisquémica se encuentra alterada en ratones knockout heterocigotos (-/+) Mn2+-SOD (150).
La tioredoxina1 (Trx1) es una proteína multifuncional de 12 kDa que limita el estrés oxidativo mediante la remoción directa de ROS y la regeneración de varias moléculas antioxidantes incluyendo ácido ascórbico, ubiquinona (Q10) y acido lipoico. Por tanto Trx1 constituye un importante mecanismo de defensa contra los ROS (151). Colectivamente, Trx1, Trx reductasa y NAD(P)H son denominados el sistema Trx, el cual opera como un potente sistema proteína-disulfuro oxidorreductasa (152). Los niveles séricos de Trx1 se encuentran elevados en pacientes con falla cardiaca y enfermedad coronaria (153). La inhibición de Trx1 en el corazón causa un incremento en el estrés oxidativo y lleva a hipertrofia cardiaca, con incrementos adicionales en estos parámetros luego de inducir sobrecarga de presión (154). Esto se correlaciona con actividad incrementada de la cascada de las MAPK ERK1/2 (154). En contraste, la sobrexpresión cardioespecífica de Trx1 reduce el estrés oxidativo y la hipertrofia cardiaca inducidos por sobrecarga de presión (154).
Aunque habría sido deseable haber evolucionado con sistemas antioxidantes más robustos, particularmente en vista de que un exceso de estrés oxidativo se encuentra en el corazón mismo de patologías degenerativas como la falla cardiaca y enfermedades neoplásicas, es posible que una actividad antioxidante excesiva pueda abolir completamente los procesos fisiológicos que requieren cantidades controladas y producción localizada de ROS. Es por esta razón que enfermedades asociadas con estrés oxidativo como la falla cardiaca (el llamado "cancer de la cardiología") y el cáncer mismo son las principales causas de morbilidad y mortalidad en la moderna era "posantibiótica" de incrementada expectativa de vida, la cual deja como resultado un creciente número en la población anciana.
Discusión, implicaciones clínicas y conclusión
Los recientes avances en el entendimiento de las fuentes de ROS y ERN en el sistema cardiovascular indican que cada uno de estos realizó un papel importante en el proceso de hipertrofia y la progresión de la falla cardiaca. Sin embargo, es posible que dentro de la fisiopatología de la falla cardiaca varios de estos sistemas interactúen y de esta forma creen círculos viciosos que se autoperpetúan llevando a una marcada alteración de la homeóstasis del sistema y daño miocárdico progresivo como consecuencia de ello.
Un escenario plausible en este sentido lo constituyen las alteraciones miocárdicas derivadas de la sobrecarga hemodinámica que ocurren en el ventrículo izquierdo como consecuencia de enfermedades comunes tales como IAM o hipertensión arterial. Esta sobrecarga hemodinámica inicia una respuesta compensadora que activa varios sistemas neuroendocrinos como el sistema adrenérgico, el sistema renina angiotensina y las endotelinas. De esta forma, actuando sobre sus receptores específicos acoplados a proteínas Gq (RAPGq), agonistas como la ET-1, noradrenalina y Ang-II activan las enzimas NOX, y en el cardiomiocito es factible que activen particularmente a Nox2 (Figura 3). Esta activación de NOX genera una parte importante del estrés oxidativo en los cardiomiocitos y otras células del tejido miocárdico (ver secciones anteriores) (155). Por su parte, la estimulación persistente de estos sistemas neuroendocrinos lleva entonces a la activación de otros sistemas generadores de ROS como la xantina oxidasa y al desacoplamiento de ONS3 (Figura 3). En favor de esta hipótesis, varios estudios han sugerido que XOR parece ser activado como consecuencia de la activación inicial de NOX2, como se expuso en las secciones previas (111, 112) (Figura 3).
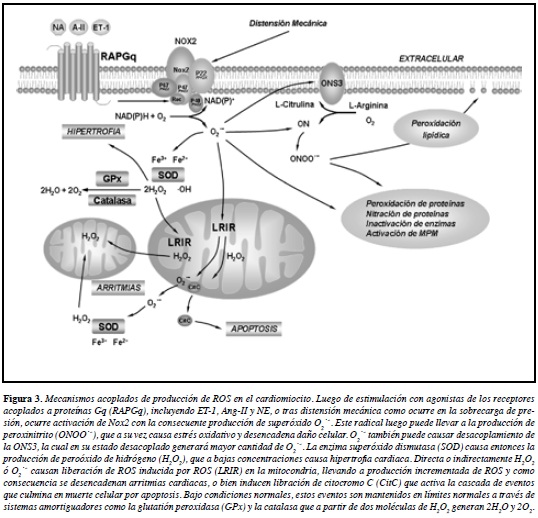
Aunque estos estímulos iniciales son importantes para mediar la hipertrofia compensadora, es factible que en etapas posteriores estos sistemas disfuncionen cada vez en forma más importante. Así, una vez que el estrés de la pared se incrementa hasta un cierto nivel, como consecuencia de la hipertrofia cardiaca compensadora, la isquemia subendocárdica puede generar condiciones de estrés para la célula suficientes como para desencadenar la muerte de los cardiomiocitos bien sea por necrosis y/o apoptosis (33-37). Esto último, aunado a la generación de estrés oxidativo derivado de NOX, XOR y el desacoplamiento de ONS3, puede llevar a alteraciones en la mitocondria de los cardiomiocitos e inducir LRIR, lo cual por un lado desencadena mayor tasa de apoptosis de cardiomiocitos y por otro lado puede generar arritmias cardiacas (Figura 3), siendo estas últimas la causa final de muerte súbita en un alto porcentaje de los pacientes con falla cardiaca (7).
Este escenario hipotético aquí planteado, podría explicar la aparente poca utilidad del uso medicamentos que interfieren con la reducción de la XOR, como el alopurinol y el oxipurinol, los cuales no han mostrado grandes beneficios sobre la mortalidad en falla cardiaca en estudios recientes (ver sección Producción de ROS por las xantina oxidasas) (87). Por el contrario, los beneficios que sobre la mortalidad en falla cardiaca tienen las propiedades antioxidantes y mejoradoras del balance nitroso-redox derivadas de la combinación de hidralazina/dinitrato de isosorbide han sido demostrados recientemente en pacientes afroamericanos en el estudio African American Heart Failure Trial (A-HeFT) (156). En esta población, la disfunción endotelial desempeña un papel muy importante en la génesis de hipertensión y falla cardiaca de origen hipertensivo (157, 158). La hidralazina es un vasodilatador que inhibe la generación de O2-; por su parte el dinitrato de isosorbide es un donante exógeno de ON. Los resultados de estudios como el A-HeFT, indican que combinaciones como ésta, mejoran la disfunción endotelial y restauran el balance nitroso/redox, con beneficios importantes sobre la mortalidad (115). Queda por discernir si esta combinación, o similares, puede tener también un impacto positivo en poblaciones con falla cardiaca diferentes a la afroamericana. Combinaciones que incluyan por ejemplo hidralazina y oxipurinol, entre otras, tienen sentido en la medida que estos fármacos bloquean diferentes aspectos de los sistemas generadores de estrés oxidativo en el sistema cardiovascular como lo muestran estudios recientes en modelos animales de hipertensión (159). Desde luego, sólo los estudios preclínicos de combinaciones de fármacos como ésta, generarán confianza suficiente del comportamiento positivo de estos fármacos in vivo como para empezar a plantear estudios en humanos.
Un blanco terapéutico potencialmente importante, sería el inhibir la NOX2, particularmente a nivel de la subunidad p47PHOX, como lo indican estudios en modelos animales (ver arriba, sección NAD(P)H oxidasas) (111). Sería deseable además, entender el sistema a tal punto de poder establecer si con sólo dirigir la terapia a un solo eslabón de esta red interactuante generadora de estrés oxidativo, se puede romper el círculo vicioso que lleva a la remodelación ventricular y progresión inexorable de la falla cardiaca. Sin embargo, dada la complejidad intrínseca del sistema es factible que aproximaciones terapéuticas dirigidas a un solo sistema no tengan el impacto positivo necesario para mejorar la mortalidad en falla cardiaca, tal como hasta ahora lo indica la evidencia disponible.
En conclusión, la falla cardiaca es un síndrome clínico severo, cuyo tratamiento farmacológico aún no logra revertir su progresión redundando en alta mortalidad y costos enormes para los sistemas de salud en todo el mundo. El estrés oxidativo/nitrosativo desempeña un papel importante en el desarrollo de hipertrofia cardiaca así como también en el proceso de remodelación ventricular que finalmente lleva al síndrome clínico de falla cardiaca. Se han hecho recientemente avances importantes en el entendimiento de los mecanismos moleculares que generan estrés oxidativo/ nitrosativo en el sistema cardiovascular. Las medidas terapéuticas empleadas en el tratamiento de la falla cardiaca actualmente (como los inhibidores del sistema renina angiotensina, antagonistas de la aldosterona o los bloqueadores β-adrenérgicos), probablemente intervienen positivamente en algunos aspectos del estrés oxidativo en la falla cardiaca. Sin embargo, estrategias terapéuticas que intervengan directa y eficazmente en la generación de ROS y ERN por parte de los sistemas aquí expuestos, podrían lograr en teoría mejorar este fenómeno e influenciar positivamente el pronóstico de la falla cardiaca. Cabe resaltar, que dada la complejidad de los diversos sistemas generadores del estrés oxidativo/ nitrosativo y su probable interacción in vivo, las estrategias terapéuticas posiblemente requieran de la combinación de fármacos a diferentes niveles de control en la producción de ROS y ERN, como se ha demostrado en la combinación de hidralazina/dinitrato de isosorbide en pacientes con falla cardiaca afro-americanos.
Referencias
1. Allender S, Scarborough P, Peto V. Coronary heart disease statistics 2007 edition. British heart foundation health promotion research group. Allender S, Scarborough P, and Peto V, editors.BHF.2007 p35-50. [ Links ]
2. Poole-Wilson PA. Cardiovascular health in Europe: a global problem. J Am Coll Cardiol 2007; 49:117-119. [ Links ]
3. Grisales H, and Bedoya GS. Change in life expectancy in connection with three large groups of causes of death in Medellin, Colombia, between 1989-1991 and 1994-1996. Pan American Journal of Public Health 2002;12: 305-12. [ Links ]
4. Lopez-Jaramillo P. Defining the research priorities to fight the burden of cardiovascular diseases in Latin America. Journal of Hypertension 2008; 26: 1886-9. [ Links ]
5. Swedberg K, Cleland J, Dargie H, Drexler H, Follath F, Komajda M, et al. Guidelines for the diagnosis and treatment of chronic heart failure: executive summary (update 2005): The Task Force for the Diagnosis and Treatment of Chronic Heart Failure of the European Society of Cardiology. Eur Heart J 2005; 26: 1115-40. [ Links ]
6. Goel A, Rakwal R, and Agrawal GK. Human Heart Failure: A Proteomics Perspective. Current Proteomics 2007; 4: 174-81. [ Links ]
7. Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, et al. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: A Report of the American College of Cardiology/ American Heart Association Task Force on Practice Guidelines (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure): Developed in Collaboration With the American College of Chest Physicians and the International Society for Heart and Lung Transplantation: Endorsed by the Heart Rhythm Society. Circulation 2005; 112: 154-235. [ Links ]
8. Katz AM. Regression of left ventricular hypertrophy: new hope for dying hearts. Circulation 1998; 98: 623-4. [ Links ]
9. Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 1996; 28: 1737-46. [ Links ]
10. Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol Heart Circ Physiol 1996; 271: 2183-9. [ Links ]
11. Moretti A, Caron L, Nakano A, Lam JT, Bernshausen A, Chen Y, et al. Multipotent Embryonic Isl1+ Progenitor Cells Lead to Cardiac, Smooth Muscle, and Endothelial Cell Diversification. Cell 2006;127(6):1151-1165. [ Links ]
12. Rota M, Padin-Iruegas ME, Misao Y, De Angelis A, Maestroni S, Ferreira- Martins J, et al. Local Activation or Implantation of Cardiac Progenitor Cells Rescues Scarred Infarcted Myocardium Improving Cardiac Function. Circ Res 2008;103(1):107-116. [ Links ]
13. Clerk A, Cullingford TE, Fuller SJ, Giraldo A, Markou T, Pikkarainen S, et al. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J Cell Physiol 2007; 212: 311-22. [ Links ]
14. Cullingford T, Markou T, Fuller S, Giraldo A, Pikkarainen S, Zoumpoulidou G, et al. Temporal regulation of expression of immediate early and second phase transcripts by endothelin-1 in cardiomyocytes. Genome Biology 2008; 9: 32. [ Links ]
15. Hoshijima M, Chien KR. Mixed signals in heart failure: cancer rules. J Clin Invest 2002;109: 849-85. [ Links ]
16. Dorn GW, II, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 2005;115: 527-37. [ Links ]
17. Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Cin Invest 1975; 56: 56-64. [ Links ]
18. Gerdes AM. Cardiac myocyte remodeling in hypertrophy and progression to failure. J Card Fail 2002; 8: 264-8. [ Links ]
19. Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. The Lancet 2006; 367: 356-67. [ Links ]
20. Meerson FZ. Compensatory Hyperfunction of the Heart and Cardiac Insufficiency. Circ Res 1962;10: 250-8. [ Links ]
21. Meerson FZ. The myocardium in hyperfunction, hypertrophy and heart failure. Circ Res 1969; 25: 1-163. [ Links ]
22. Katz AM . Energy requirements of contraction and relaxation: Implications of inotropic stimulation of the failing heart. In Inotropic Stimulation and Myocardial Energetics. Just H, Holubarsch C, and Scholz H, editors.New York: Springer- Verlag.1989 p49. [ Links ]
23. Colucci WS. Molecular and Cellular Mechanisms of Myocardial Failure. Am J Cardiol 1997; 80: 15-25. [ Links ]
24. Hunter JJ, Chien KR. Signaling pathways for cardiac hypertrophy and failure. N Engl J Med 1999;341(17):1276-1283. [ Links ]
25. Katz AM. Maladaptive Growth in the Failing Heart: The Cardiomyopathy of Overload. Cardiovascular Drugs and Therapy 2002;16: 245-9. [ Links ]
26. Pfeffer J, Pfeffer M, Fletcher P, Braunwald E. Alterations of cardiac performance in rats with established spontaneous hypertension. Am J Cardiol 1979; 44: 994-8. [ Links ]
27. Pfeffer JM, Pfeffer MA, Fishbein MC, Frohlich ED. Cardiac function and morphology with aging in the spontaneously hypertensive rat. Am J Physiol 1979; 237: 461-8. [ Links ]
28. Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, et al. Myocardial infarct size and ventricular function in rats. Circ Res 1979; 44: 503-12. [ Links ]
29. Pfeffer MA, Pfeffer JM, Steinberg C, Finn P. Survival after an experimental myocardial infarction: beneficial effects of long-term therapy with captopril. Circulation 1985; 72: 406-12. [ Links ]
30. Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, et al. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem 1993; 268: 18532-41. [ Links ]
31. Becker LB, vanden Hoek TL, Shao ZH, Li CQ, Schumacker PT. Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am J Physiol Heart Circ Physiol 1999; 277: 2240-6. [ Links ]
32. Du G, Mouithys-Mickalad A, Sluse FE. Generation of superoxide anion by mitochondria and impairment of their functions during anoxia and reoxygenation in vitro. Free Radic Biol Med 1998; 25: 1066-74. [ Links ]
33. Vatner SF. Reduced subendocardial myocardial perfusion as one mechanism for congestive heart failure. Am J Cardiol 1988; 62: 94-98. [ Links ]
34. Vatner SF, Shannon R, Hittinger L. Reduced subendocardial coronary reserve. A potential mechanism for impaired diastolic function in the hypertrophied and failing heart. Circulation 1990; 81: 1118-14. [ Links ]
35. Vatner SF, and Hittinger L. Coronary vascular mechanisms involved in decompensation from hypertrophy to heart failure. J Am Coll Cardiol 1993; 22: 34-40. [ Links ]
36. Helmer GA, McKirnan MD, Shabetai R, Boss GR, Ross J, Hammond HK. Regional Deficits of Myocardial Blood Flow and Function in Left Ventricular PacingûInduced Heart Failure. Circulation 1996; 94: 2260-7. [ Links ]
37. Davies JE, Whinnett ZI, Francis DP, Manisty CH, guado-Sierra J, Parker KH et al. Evidence of a Dominant Backward-Propagating "Suction" Wave Responsible for Diastolic Coronary Filling in Humans, Attenuated in Left Ventricular Hypertrophy. Circulation 2006; 113: 1768-78. [ Links ]
37. Davies JE, Whinnett ZI, Francis DP, Manisty CH, guado-Sierra J, Parker KH et al. Evidence of a Dominant Backward-Propagating "Suction" Wave Responsible for Diastolic Coronary Filling in Humans, Attenuated in Left Ventricular Hypertrophy. Circulation 2006; 113: 1768-78. [ Links ]
38. Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep 1997; 17: 3-8. [ Links ]
39. Gnaiger E, Kuznetsov AV. Mitochondrial respiration at low levels of oxygen and cytochrome c. Biochem Soc Trans 2002; 30: 252-8. [ Links ]
40. Barth E, Stammler G, Speiser B, and Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 1992; 24: 669-81. [ Links ]
41. Ayettey AS TCYRD. Ultrastructural characteristics of atrial, ventricular and subendocardial (Purkinje) cells of the fruit-eating bat Eidolon helvum. Acta Anat (Basel) 1993; 147: 89-96. [ Links ]
42. Davies KJ. Oxidative stress: the paradox of aerobic life. Biochem Soc Symp 1995; 61: 611-31. [ Links ]
43. Raha S, and Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci 2000; 25: 502-8. [ Links ]
44. Genova ML, Pich MM, Biondi A, Bernacchia A, Falasca A, Bovina C, et al. Production of Oxygen Radical Species and the Role of Coenzyme Q as an Antioxidant. Exp Biol Med 2003; 228: 506-13. [ Links ]
45. Kinnula VL, Crapo JD. Superoxide dismutases in malignant cells and human tumors. Free Radic Biol Med 2004; 36: 718-44. [ Links ]
46. Nohl H, Jordan W. The metabolic fate of mitochondrial hydrogen peroxide. Eur J Biochem 1980; 111: 203-10. [ Links ]
47. Halliwell B. Tell me about free radicals, doctor: a review. J R Soc Med 1989; 82: 747-52. [ Links ]
48. Schroeder E, Ponting CP. Evidence that peroxiredoxins are novel members of the thioredoxin fold superfamily. Protein Sci 1998; 7: 2465-8. [ Links ]
49. Vasquez-Vivar J, Kalyanaraman B, Kennedy MC. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J Biol Chem 2000; 275: 14064-9. [ Links ]
50. Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev 1979; 59: 527-605. [ Links ]
51. Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, et al. Mitochondrial Electron Transport Complex I Is a Potential Source of Oxygen Free Radicals in the Failing Myocardium. Circ Res 1999;85(4):357-363. [ Links ]
52. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 2002; 277: 44784-90. [ Links ]
53. Ide T, Tsutsui H, Kinugawa S, Suematsu N, Hayashidani S, Ichikawa K, et al. Direct Evidence for Increased Hydroxyl Radicals Originating From Superoxide in the Failing Myocardium. Circ Res 2000; 86:152-7. [ Links ]
54. Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura Ki, et al. Mitochondrial DNA Damage and Dysfunction Associated With Oxidative Stress in Failing Hearts After Myocardial Infarction. Circ Res 2001; 88: 529-35. [ Links ]
55. Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, Ide T, Hayashidani S, et al. Oxidative Stress Mediates Tumor Necrosis Factor-{alpha}-Induced Mitochondrial DNA Damage and Dysfunction in Cardiac Myocytes. Circulation 2003; 107: 1418- 23. [ Links ]
56. Pletjushkina OY, Fetisova EK, Lyamzaev KG, Ivanova OY, Domnina LV, Vyssokikh MY, et al. Long-distance apoptotic killing of cells is mediated by hydrogen peroxide in a mitochondrial ROS-dependent fashion. Cell Death Differ 2005;12: 1442-4. [ Links ]
57. Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochem J 1972;128: 617-630. [ Links ]
58. Nieminen AL, Byrne AM, Herman B, Lemasters JJ. Mitochondrial permeability transition in hepatocytes induced by t-BuOOH: NAD(P)H and reactive oxygen species. Am J Physiol 1997; 272: 1286-94. [ Links ]
59. Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000; 192: 1001-14. [ Links ]
60. Leach JK, Black SM, Schmidt-Ullrich RK, Mikkelsen RB. Activation of constitutive nitric-oxide synthase activity is an early signaling event induced by ionizing radiation. J Biol Chem 2002; 277: 15400-6. [ Links ]
61. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta 2006; 1757: 509-17. [ Links ]
62. Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie 2002; 84: 153-166. [ Links ]
63. Crompton M, Barksby E, Johnson N, Capano M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie 2002; 84: 143-52. [ Links ]
64. Galiegue S, Tinel N, Casellas P. The peripheral benzodiazepine receptor: a promising therapeutic drug target. Curr Med Chem 2003; 10: 1563-72. [ Links ]
65. Shanmuganathan S, Hausenloy DJ, Duchen MR, Yellon DM. Mitochondrial permeability transition pore as a target for cardioprotection in the human heart. Am J Physiol Heart Circ Physiol 2005; 289: 237-42. [ Links ]
66. Davidson SM, Hausenloy D, Duchen MR, Yellon DM. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int J Biochem Cell Biol 2006; 38: 414-419. [ Links ]
67. Akar FG, Aon MA, Tomaselli GF, O'Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest 2005;115: 3527-35. [ Links ]
68. Lakatta EG, Sollott SJ. The "Heartbreak" of Older Age. Mol Interv 2002; 2: 431-446. [ Links ]
69. Aon MA, Cortassa S, Akar FG, O'Rourke B. Mitochondrial criticality: a new concept at the turning point of life or death. Biochim Biophys Acta 2006; 1762: 232-40. [ Links ]
70. Kwon SH, Pimentel DR, Remondino A, Sawyer DB, Colucci WS. H2O2 regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. Journal of Molecular and Cellular Cardiology 2003; 35: 615-21. [ Links ]
71. Cook SA, Sugden PH, Clerk A. Regulation of bcl-2 family proteins during development and in response to oxidative stress in cardiac myocytes: association with changes in mitochondrial membrane potential. Circ Res 1999; 85: 940-9. [ Links ]
72. Kang YJ, Zhou ZX, Wang GW, Buridi A, Klein JB. Suppression by Metallothionein of Doxorubicin-induced Cardiomyocyte Apoptosis through Inhibition of p38 Mitogen-activated Protein Kinases. J Biol Chem 2000; 275: 13690-8. [ Links ]
73. Stirpe F, and la Corte E. The regulation of rat liver xanthine oxidase. Conversion in vitro of the enzyme activity from dehydrogenase (type D) to oxidase (type O). J Biol Chem 1969; 244: 3855-33. [ Links ]
74. Waud WR, Rajagopalan KV. Purification and properties of the NAD+-dependent (type D) and O2-dependent (type O) forms of rat liver xanthine dehydrogenase. Arch Biochem Biophys 1976; 172: 354-64. [ Links ]
75. Olson JS, Ballou DP, Palmer G, Massey V. The Mechanism of Action of Xanthine Oxidase. J Biol Chem 1974; 249: 4363-82. [ Links ]
76. Hille R, Massey V. Studies on the oxidative half-reaction of xanthine oxidase. J Biol Chem 1981; 256: 9090-95. [ Links ]
77. Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, et al. Allopurinol Improves Myocardial Efficiency in Patients With Idiopathic Dilated Cardiomyopathy. Circulation 2001; 104: 2407-2411. [ Links ]
78. Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SVY, Tejani AD, et al. From the Cover: Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. PNAS 2004; 101: 15944-8. [ Links ]
79. Ukai T, Cheng CP, Tachibana H, Igawa A, Zhang ZS, Cheng HJ, et al. Allopurinol Enhances the Contractile Response to Dobutamine and Exercise in Dogs With Pacing-Induced Heart Failure. Circulation 2001; 103: 750-5. [ Links ]
80. Ekelund UEG, Harrison RW, Shokek O, Thakkar RN, Tunin RS, Senzaki H, et al. Intravenous Allopurinol Decreases Myocardial Oxygen Consumption and Increases Mechanical Efficiency in Dogs With Pacing-Induced Heart Failure. Circ Res 1999; 85: 437-45. [ Links ]
81. Engberding N, Spiekermann S, Schaefer A, Heineke A, Wiencke A, Muller M, et al. Allopurinol Attenuates Left Ventricular Remodeling and Dysfunction After Experimental Myocardial Infarction: A New Action for an Old Drug?. Circulation 2004;110(15):2175-2179. [ Links ]
82. de Jong JW, Schoemaker RG, de Jonge R, Bernocchi P, Keijzer E, Harrison R, et al. Enhanced Expression and Activity of Xanthine Oxidoreductase in the Failing Heart. Journal of Molecular and Cellular Cardiology 2000; 32: 2083-9. [ Links ]
83. Minhas KM, Saraiva RM, Schuleri KH, Lehrke S, Zheng M, Saliaris AP, et al. Xanthine Oxidoreductase Inhibition Causes Reverse Remodeling in Rats With Dilated Cardiomyopathy. Circ Res 2006; 98: 271-9. [ Links ]
84. Farquharson CAJ, Butler R, Hill A, Belch JJF, Struthers AD. Allopurinol Improves Endothelial Dysfunction in Chronic Heart Failure. Circulation 2002; 106: 221-6. [ Links ]
85. Struthers AD, Donnan PT, Lindsay P, McNaughton D, Broomhall J, MacDonald TM, et al. Effect of allopurinol on mortality and hospitalisations in chronic heart failure: a retrospective cohort study. Heart 2002; 87: 229-34. [ Links ]
86. George J, Carr E, Davies J, Belch JJF, Struthers A. High-Dose Allopurinol Improves Endothelial Function by Profoundly Reducing Vascular Oxidative Stress and Not by Lowering Uric Acid. Circulation 2006; 114: 2508-16. [ Links ]
87. Hare JM, Mangal B, Brown J, Fisher Jr C, Freudenberger R, Colucci WS, et al. Impact of Oxypurinol in Patients With Symptomatic Heart Failure: Results of the OPT-CHF Study. J Am Coll Cardiol 2008; 51: 2301-9. [ Links ]
88. Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 2004; 4: 181-9. [ Links ]
89. Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem 2004; 279: 45935-41. [ Links ]
90. Martyn KD, Frederick LM, von LK, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal 2006;18: 69-82. [ Links ]
91. Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 2000;86(5):494-501. [ Links ]
92. Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH Oxidase During Progression of Cardiac Hypertrophy to Failure. Hypertension 2002; 40: 477-84. [ Links ]
93. Xiao L, Pimentel DR, Wang J, Singh K, Colucci WS, Sawyer DB. Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol 2002; 282: 926-34. [ Links ]
94. Higashi M, Shimokawa H, Hattori T, Hiroki J, Mukai Y, Morikawa K, et al. Long-Term Inhibition of Rho-Kinase Suppresses Angiotensin II-Induced Cardiovascular Hypertrophy in Rats In Vivo: Effect on Endothelial NAD(P)H Oxidase System. Circ Res 2003; 93: 767-75. [ Links ]
95. Colston JT, de la Rosa SD, Strader JR, Anderson MA, and Freeman GL. H2O2 activates Nox4 through PLA2-dependent arachidonic acid production in adult cardiac fibroblasts. FEBS Letters 2005; 579: 2533-40. [ Links ]
96. Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, et al. NAD(P)H Oxidase 4 Mediates Transforming Growth Factor-{beta}1-Induced Differentiation of Cardiac Fibroblasts Into Myofibroblasts. Circ Res 2005; 97: 900-7. [ Links ]
97. Li J, Stouffs M, Serrander L, Banfi B, Bettiol E, Charnay Y, et al. The NADPH Oxidase NOX4 Drives Cardiac Differentiation: Role in Regulating Cardiac Transcription Factors and MAP Kinase Activation. Mol Biol Cell 2006;17: 3978-88. [ Links ]
98. Sabri A, Hughie HH, and Lucchesi PA. Regulation of Hypertrophic and Apoptotic Signaling Pathways by Reactive Oxygen Species in Cardiac Myocytes. Antioxidants & Redox Signaling 2003; 5: 731-40. [ Links ]
99. Wenzel S, Taimor G, Piper HM, Schl³ter KD. Redox-sensitve intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-b expression in adult ventricular cardiomyocytes. FASEB J 200100- 0827fje. [ Links ]
100. Kinugawa S, Zhang J, Messina E, Walsh E, Huang H, Kaminski PM, et al. gp91phox-containing NAD(P)H oxidase mediates attenuation of nitric oxidedependent control of myocardial oxygen consumption by ANG II. Am J Physiol Heart Circ Physiol 2005;289(2):H862-H867. [ Links ]
101. Krijnen PAJ, Meischl C, Hack CE, Meijer CJLM, Visser CA, Roos D, and Niessen HWM. Increased Nox2 expression in human cardiomyocytes after acute myocardial infarction. J Clin Pathol 2003; 56: 194-199. [ Links ]
102. Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G, et al. Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 2003; 41: 2164-71. [ Links ]
103. Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal Role of a gp91phox-Containing NADPH Oxidase in Angiotensin II-Induced Cardiac Hypertrophy in Mice. Circulation 2002; 105: 293-6. [ Links ]
104. Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, et al. Contrasting Roles of NADPH Oxidase Isoforms in Pressure-Overload Versus Angiotensin II-Induced Cardiac Hypertrophy. Circ Res 2003; 93: 802-5. [ Links ]
105. Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC, et al. Involvement of the Nicotinamide Adenosine Dinucleotide Phosphate Oxidase Isoform Nox2 in Cardiac Contractile Dysfunction Occurring in Response to Pressure Overload. J Am Coll Cardiol 2006; 47: 817-26. [ Links ]
106. Matsushita H, Lee KH, Tsao PS. Cyclic strain induces reactive oxygen species production via an endothelial NAD(P)H oxidase. J Cell Biochem 2001; 36: 3699-106. [ Links ]
107. Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, et al. Li H, Bodenschatz M, August M, Kleschyov AL et al. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res 2002; 90: 58-65. [ Links ]
108. Lund AK, Peterson SL, Timmins GS, Walker MK. Endothelin-1-Mediated Increase in Reactive Oxygen Species and NADPH Oxidase Activity in Hearts of Aryl Hydrocarbon Receptor (AhR) Null Mice. Toxicol Sci 2005; 88: 265-73. [ Links ]
109. Amin JK, Xiao L, Pimental DR, Pagano PJ, Singh K, Sawyer DB, et al. Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J Mol Cell Cardiol 2001; 33: 131-9. [ Links ]
110. Li JM, Shah AM. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J Biol Chem 2003; 278:12094-100. [ Links ]
111. Doerries C, Grote K, Hilfiker-Kleiner D, Luchtefeld M, Schaefer A, Holland SM, et al. Critical Role of the NAD(P)H Oxidase Subunit p47phox for Left Ventricular Remodeling/Dysfunction and Survival After Myocardial Infarction. Circ Res 2007; 100: 894-903. [ Links ]
112. McNally JS, Saxena A, Cai H, Dikalov S, Harrison DG. Regulation of xanthine oxidoreductase protein expression by hydrogen peroxide and calcium. Arterioscler Thromb Vasc Biol 2005; 25:1623-8. [ Links ]
113. Saraiva RM, and Hare JM. Nitric oxide signaling in the cardiovascular system: implications for heart failure. Curr Opin Cardiol 2006; 21: 221-8. [ Links ]
114. Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 2007; 93: 903-7. [ Links ]
115. Hare JM. Nitroso-redox balance in the cardiovascular system. N Engl J Med 2004; 351: 2112-4. [ Links ]
116. Hare JM, Stamler JS. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest 2005; 115: 509-17. [ Links ]
117. Champion HC, Georgakopoulos D, Takimoto E, Isoda T, Wang Y, Kass DA. Modulation of in vivo cardiac function by myocyte-specific nitric oxide synthase-3. Circ Res 2004; 94: 657-63. [ Links ]
118. Wollert KC, Fiedler B, Gambaryan S, Smolenski A, Heineke J, Butt E, et al. Gene transfer of cGMP-dependent protein kinase I enhances the antihypertrophic effects of nitric oxide in cardiomyocytes. Hypertension 2002; 39: 87-92. [ Links ]
119. Wollert KC, Drexler H. Regulation of cardiac remodeling by nitric oxide: focus on cardiac myocyte hypertrophy and apoptosis. Heart Fail Rev 2002; 7: 317-25. [ Links ]
120. Horio T, Nishikimi T, Yoshihara F, Matsuo H, Takishita S, Kangawa K. Inhibitory regulation of hypertrophy by endogenous atrial natriuretic peptide in cultured cardiac myocytes. Hypertension 2000; 35: 19-24. [ Links ]
121. Kishimoto I, Rossi K, Garbers DL. A genetic model provides evidence that the receptor for atrial natriuretic peptide (guanylyl cyclase-A) inhibits cardiac ventricular myocyte hypertrophy. Proc Natl Acad Sci U S A 2001; 98: 2703-6. [ Links ]
122. Fiedler B, Lohmann SM, Smolenski A, Linnemuller S, Pieske B, Schroder F, et al. Inhibition of calcineurin-NFAT hypertrophy signaling by cGMP-dependent protein kinase type I in cardiac myocytes. Proc Natl Acad Sci U S A 2002; 99: 11363-8. [ Links ]
123. Munzel T, Daiber A, Ullrich V, Mulsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol 2005; 25: 1551-7. [ Links ]
124. Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem 1998; 273: 25804-8. [ Links ]
125. Xia Y. Superoxide generation from nitric oxide synthases. Antioxid Redox Signal 2007; 9: 1773-8. [ Links ]
126. Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J 2001; 357: 593-615. [ Links ]
127. Ghosh DK, Salerno JC. Nitric oxide synthases: domain structure and alignment in enzyme function and control. Front Biosci 2003: 193-209. [ Links ]
128. Ghosh DK, Holliday MA, Thomas C, Weinberg JB, Smith SM, Salerno JC. Nitric-oxide synthase output state. Design and properties of nitric-oxide synthase oxygenase/FMN domain constructs. J Biol Chem 2006; 281: 14173-83. [ Links ]
129. Rosen GM, Tsai P, Weaver J, Porasuphatana S, Roman LJ, Starkov AA, et al. The role of tetrahydrobiopterin in the regulation of neuronal nitric-oxide synthase-generated superoxide. J Biol Chem 2002; 277: 40275-80. [ Links ]
130. Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem 2003;278(25):22546-22554. [ Links ]
131. Maier W, Cosentino F, Lutolf RB, Fleisch M, Seiler C, Hess OM, et al. Tetrahydrobiopterin improves endothelial function in patients with coronary artery disease. J Cardiovasc Pharmacol 2000; 35: 173-8. [ Links ]
132. Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol 2004; 24: 413-20. [ Links ]
133. Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, et al. Oxidant stress from nitric oxide synthase–3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Inves 2005; 115: 1221-31. [ Links ]
134. Siwik DA, Pagano PJ, and Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol 2001; 280: C53-C60. [ Links ]
135. Kameda K, Matsunaga T, Abe N, Hanada H, Ishizaka H, Ono H, Saitoh M, et al. Correlation of oxidative stress with activity of matrix metalloproteinase in patients with coronary artery disease. Possible role for left ventricular remodelling. Eur Heart J 2003; 24: 2180-5. [ Links ]
136. Mori S, Gibson G, McTiernan CF. Differential expression of MMPs and TIMPs in moderate and severe heart failure in a transgenic model. J Card Fail 2006; 12: 314-25. [ Links ]
137. Yan AT, Yan RT, Spinale FG, Afzal R, Gunasinghe HR, Arnold M, et al. Plasma matrix metalloproteinase-9 level is correlated with left ventricular volumes and ejection fraction in patients with heart failure. J Card Fail 2006; 12: 514-9. [ Links ]
138. Henderson BC, Tyagi N, Ovechkin A, Kartha GK, Moshal KS, and Tyagi SC. Oxidative remodeling in pressure overload induced chronic heart failure. Eur J Heart Fail 2007; 9: 450-7. [ Links ]
139. Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, et al. Ketner EA, Majmudar M, Gabrielson K, Halushka MK et al. Reversal of Cardiac Hypertrophy and Fibrosis From Pressure Overload by Tetrahydrobiopterin: Efficacy of Recoupling Nitric Oxide Synthase as a Therapeutic Strategy. Circulation 2008; 117: 2626-6. [ Links ]
140. Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest 2002; 109: 817-26. [ Links ]
141. Bendall JK, Alp NJ, Warrick N, Cai S, Adlam D, Rockett K, et al. Yokoyama M, Kawashima S, and Channon KM. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res 2005; 97: 864-71. [ Links ]
142. Scherrer-Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT, et al. Endothelial Nitric Oxide Synthase Limits Left Ventricular Remodeling After Myocardial Infarction in Mice. Circulation 2001; 104:1286-91. [ Links ]
143. Ruetten H, Dimmeler S, Gehring D, Ihling C, Zeiher AM. Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovasc Res 2005; 66: 444-53. [ Links ]
144. Ichinose F, Bloch KD, Wu JC, Hataishi R, Aretz HT, Picard MH, et al. Pressure overload-induced LV hypertrophy and dysfunction in mice are exacerbated by congenital NOS3 deficiency. Am J Physiol Heart Circ Physiol 2004; 286: 1070-5. [ Links ]
145. Buys ES, Raher MJ, Blake SL, Neilan TG, Graveline AR, Passeri JJ, et al. Cardiomyocyte-restricted restoration of nitric oxide synthase 3 attenuates left ventricular remodeling after chronic pressure overload. Am J Physiol Heart Circ Physiol 2007; 293: 620-7. [ Links ]
146. Ohta H, Adachi T, and Hirano K. Internalization of human extracellularsuperoxide dismutase by bovine aortic endothelial cells. Free Radical Biology and Medicine 1994; 16: 501-7. [ Links ]
147. Dhalla NS, Elmoselhi AB, Hata T, Makino N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovascular Research 2000; 47: 446-56. [ Links ]
148. Shiomi T, Tsutsui H, Matsusaka H, Murakami K, Hayashidani S, Ikeuchi M, Wen J, Kubota T, Utsumi H, and Takeshita A. Overexpression of Glutathione Peroxidase Prevents Left Ventricular Remodeling and Failure After Myocardial Infarction in Mice. Circulation 2004; 109: 544-9. [ Links ]
149. Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, Chua BH. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol 1998; 30: 2281-9. [ Links ]
150. Asimakis GK, Lick S, Patterson C. Postischemic Recovery of Contractile Function is Impaired in SOD2+/- but Not SOD1+/- Mouse Hearts. Circulation 2002; 105: 981-6. [ Links ]
151. Yamawaki H, Haendeler J, Berk BC. Thioredoxin: A Key Regulator of Cardiovascular Homeostasis. Circ Res 2003; 93: 1029-33. [ Links ]
152. Masutani H, Yodoi J. Thioredoxin: Overview. In Methods in Enzymology: Protein Sensors and Reactive Oxygen Species Part A. Helmut Sies and Lester Packer, editor. Academic Press 2002 p.279-286. [ Links ]
153. Kishimoto C, Shioji K, Nakamura H, Nakayama Y, Yodoi J, et al. Serum thioredoxin (TRX) levels in patients with heart failure. Jpn Circ J 2001; 65: 491-4. [ Links ]
154. Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, et al. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J Clin Invest 2003; 112: 1395-406. [ Links ]
155. Callera GE, Tostes RC, Yogi A, Montezano ACI, Touyz RM. Endothelin-1- induced oxidative stress in DOCA-salt hypertension involves NADPH-oxidaseindependent mechanisms. Clin Sci 2006; 110: 243-53. [ Links ]
156. Taylor AL, Ziesche S, Yancy C, Carson P, D'Agostino R, Jr., Ferdinand K, et al. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med 2004; 351: 2049-57. [ Links ]
157. Lapu-Bula R, Ofili E, Oduwole A, Lankford B, Pack C, St Vrain J, et al. Relation of blood pressure to endothelium-dependent vasodilation in African American normotensives and hypertensives. American Journal of Hypertension 2001; 14: 199. [ Links ]
158. Li R, Lyn D, Lapu-Bula R, Oduwole A, Igho-Pemu P, Lankford B, Morgan J, et al. Relation of endothelial nitric oxide synthase gene to plasma nitric oxide level, endothelial function, and blood pressure in African Americans. American Journal of Hypertension 2004; 17: 560-7. [ Links ]
159. Viel EC, Benkirane K, Javeshghani D, Touyz RM, Schiffrin EL. Xanthine oxidase and mitochondria contribute to vascular superoxide anion generation in DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol 2008; 295: 281-8. [ Links ]

