










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.35 no.2 Bogotá Apr./June 2010
PRESENTACIÓN DE CASOS
Hypocalcemia due to vitamin D dependency type I in a patient with uric acid nephropathy
César A. Restrepo(1), Angela María Benjumea(2) • Manizales (Caldas)
(1) Internista Nefrólogo de la Universidad de Caldas, Profesor Asociado de la Universidad de Caldas.
(2) Médico Cirujano Universidad de Caldas, Residente segundo año Medicina Interna y Geriatría Universidad de Caldas.
Correspondencia. Dr. Cesar A. Restrepo V. Carrera 28B # 71ª56 Edificio los Olivos tercer piso. Teléfonos: 8871572 y 8783050. Fax: 8876692.Manizales (Caldas)
E-mail: caugustorv@une.net.co
Recibido: 10/XI/09 Aceptado: 21/IV/10
Resumen
Se discute el caso de un paciente hombre de 37 años con hiperuricemia, nefrolitiasis múltiple bilateral, y trastorno metabólico mineral caracterizado por hipocalcemia, hipofosfatemia, hipocalciuria y normofosfaturia de 10 años de evolución, con pobre respuesta al manejo médico instaurado y sin diagnóstico preciso. Tras análisis clínicos y apoyo en exámenes de laboratorio se evidencia dependencia tipo I a la vitamina D (Acta Med Colomb 2010; 35: 96-99).
Palabras clave: hipocalcemia, vitamina D, litiasis renal.
Abstract
We discuss the case of a 37-year-old man with hyperuricemia, bilateral nephrolithiasis, and mineral metabolic disorder characterized by hypocalcemia, hypophosphatemia, and hypocalciuria without phosphaturia. The diseased had developed over 10 years without an accurate diagnosis, and had responded poorly to medical management. After clinical analyses and laboratory workup, vitamin D dependency type I was diagnosed (Acta Med Colomb 2010; 35: 96-99).
Keywords: hypocalcemia, vitamin D, nephrolithiasis.
Introducción
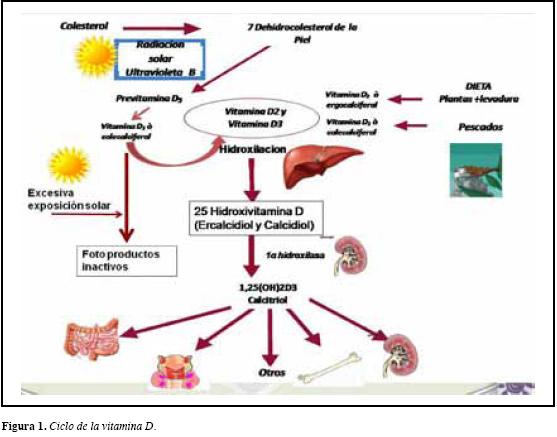
Los trastornos del metabolismo óseo y mineral en algunas situaciones son de difícil diagnóstico, principalmente para los médicos que no están familiarizados con las hormonas que intervienen en su regulación. La vitamina D en su forma activa es requerida para optimizar los procesos de absorción intestinal de calcio y fósforo, inducir a nivel óseo maduración de osteoclastos, y frenar la producción de paratohormona (Figura 1). Trastornos en diversas etapas del ciclo de la vitamina D pueden dar lugar a alteraciones electrolíticas, principalmente manifestadas en modificaciones en los niveles séricos de calcio y fósforo con sus respectivas repercusiones óseas y sistémicas (1). La hiperuricemia es una alteración metabólica relativamente común, y porta complicaciones sistémicas y renales de consideración. A continuación presentamos el caso clínico complejo de un paciente que reúne alteraciones en el metabolismo óseo y del ácido úrico.
Descripcion del caso
Hombre de 37 años residente en zona urbana, comerciante agropecuario, exposición solar frecuente, con cuadro clínico que se inicia 10 años previos a la evaluación por nefrología, consistente en dolor en flanco, disuria, hematuria, detectándose por ecografía renal y radiografía simple de abdomen nefrolitiasis múltiple bilateral por cálculos radio opacos; en otros paraclínicos se evidenció como datos importantes hiperuricemia, hiperazoemia, hipocalcemia, hipofosfatemia, hipocalciuria, normofosfaturia, T4 libre, TSH y calcitonina normales y una PTH intacta de 77,6 (vn: 15-65). Se solicito ecografía de cuello y se detectó adenoma paratiroideo derecho, llevándose el paciente posteriormente a resección quirúrgica del mismo. El reporte de la pieza anatómica fue compatible con adenoma de células principales (paratiroideo). En los cuatro años siguientes el paciente continuó presentando síntomas característicos de litiasis renal y de vías urinarias, detectándose siempre en los paraclínicos hiperuricemia, hipocalcemia, normoalbuminemia, normohipofosfatemia, hipocalciuria, normofosfaturia con valores de PTH intacta en rangos normales (37.6- 42.8), decidiéndose por endocrinología iniciarle calcitriol (vitamina D activa o 1,25(OH)vitamina D3) 0,50 mcg por día y allopurinol 100 mg por día.
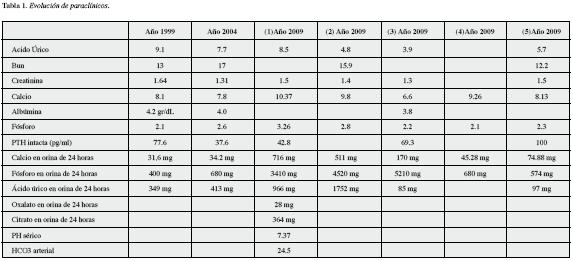
Con la terapia anterior es interconsultado a nefrología a los 10 años del inicio de los síntomas por presentar persistencia de crisis de urolitiasis y deterioro de la función renal. En los paraclínicos solicitados se detectó elevación de azoados, hiperuricemia, normofosfatemia, hipercalcemia, hipercalciuria, hiperfosfaturia e hiperuricosuria, la PTH intacta se detectó en rangos normales. Se consideró que el paciente podría estar cursando con tubulopatía proximal, pero sus gases arteriales fueron normales, lo cual orientó hacia intoxicación por vitamina D, razón por la que se suspendió el calcitriol y se aumentó la dosis de alopurinol a 300 mg por día. Treinta días más tarde sus valores de ácido úrico, calcio y fósforo sérico se normalizaron, al igual que la excreción urinaria de calcio y fósforo. En control a los 60 días, de nuevo el paciente desarrolló hipocalcemia, normoalbuminemia e hipofosfatemia con elevación de la PTH intacta a valores por arriba del rango de la normalidad, con descenso en la eliminación del calcio urinario y aumento en la de fosfatos. Se solicitaron niveles de vitamina D (25 Hidroxicalciferol) reportándose en 27 ng/mL, optándose por reiniciarse el calcitriol a dosis de 0.25 mcg día de por medio. En los controles posteriores practicados a los 90 y 120 días el paciente presenta cifras altas de PTH intacta, normales de ácido úrico, levemente bajas de calcio y fósforo séricos, valores de calcio urinarios continúan bajos, y los de fósforo regresaron a la normalidad (Tabla 1). Se inició carbonato de calcio 600 mg lejos de las comidas (para evitar su efecto quelante de fosfatos), y se está en proceso de alcanzar la dosis óptima de calcitriol.
Discusión
La hiperuricemia se define por una concentración sérica de ácido úrico mayor a 6.8 mg/dl, se presenta en 2%-13,2% de los adultos ambulatorios, y en un porcentaje mayor en pacientes hospitalizados (2). Afecta principalmente a los hombres, y principalmente se hace evidente en la pubertad; las mujeres en etapa reproductiva logran eliminar concentraciones mayores de ácido úrico al inhibir su reabsorción tubular, efecto generado por los estrógenos. Entre la principales complicaciones de la hiperuricemia está la gota, y a nivel renal, nefro-urolitiasis, nefropatía crónica por uratos y nefropatía aguda por ácido úrico. Es el resultado de dos opciones: 1) sobreproducción de uratos y 2) disminución en la eliminación de ácido úrico. La última opción explica el 85%-90% de los casos, siendo el resultado de trastornos en la filtración glomerular, baja secreción tubular de uratos o aumento en su reabsorción. La sobreproducción de uratos puede ser el resultado de dietas altas en purinas o aumento en la producción endógena de las mismas.
El algoritmo diagnóstico de la hiperuricemia (Figura 2) está dirigido precisamente a identificar en qué grupo se encuentra el paciente, puesto que si es un sobre productor de uratos se beneficiaría de la utilización de alopurinol o febuxostat, inhibidores de la xantin-oxidasa; y en casos especiales, de uricasa intravenosa, enzima que convierte el ácido úrico a un compuesto más soluble, la alantoína. Si el paciente en cambio es un bajo excretor; requerirá la administración de uricosúricos como el probenecid, sulfinpirazona u otros con menor potencia como el losartán, fenofibrato y vitamina C (3-5).
En nuestro paciente el valor de excreción de ácido úrico en orina de 24 horas fue de 349 miligramos, lo cual lo ubica en el grupo de bajos excretores; sin embargo la presencia de nefro-urolitiasis contraindica la utilización de uricosúricos, por lo que se optó por utilizar alopurinol, siendo importante resaltar que a medida que se vaya disminuyendo su función renal, éste se acumula generando riesgos de toxicidad, por lo que sería necesario ajustar su dosis o acudir a la utilización de febuxostat, el cual no requiere reajuste de dosis a bajas tasas de filtración glomerular.
En cuanto al trastorno metabólico mineral, nos encontramos frente a un paciente con hipocalcemia verdadera (sin hipoalbuminemia), con fósforo normal a bajo y niveles séricos normales-altos de hormona paratiroidea intacta (PTHi). Existen diversos enfoques para analizar un paciente con hipocalcemia, pero consideramos que el propuesto por el panel de expertos del ministerio de la salud, trabajo y bienestar japonés es el más aplicable a nuestro paciente y es el que utilizaremos (Figura 3) (6).
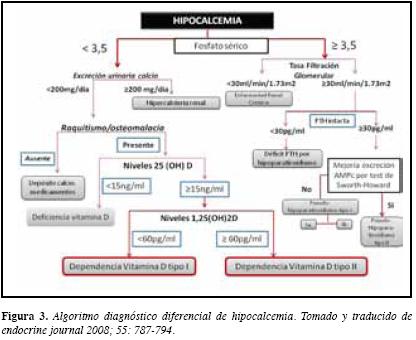
Aplicando el algoritmo recomendado por el anterior panel de expertos, se aprecia que la excreción de calcio urinaria del paciente es menor de 200 mg por día, los niveles de 25(OH) D son mayores de 15 ng/mL, con lo cual sólo queda como probable diagnóstico en nuestro paciente dependencia a la vitamina D, descartándose deficiencia de vitamina D. Esta entidad se caracteriza por requerir de por vida la administración de vitamina D, de ahí su nombre. Se han identificado dos variedades de dependencia a la vitamina D:
El tipo II (conocido también como raquitismo hipocalcémico resistente a la vitamina D), desorden autosómico dominante cuya característica es la presencia de mutaciones en el gen del receptor para la vitamina D3 activa, con lo cual aunque se forma adecuadamente la 1,25(OH)2 vitamina D3, no puede ser reconocida por su receptor, detectándose niveles séricos altos de la misma. Un pequeño grupo de pacientes responden al tratamiento con altas dosis de calcitriol, pero otros no, con lo cual se establece que existe la presencia de diversas alteraciones en el receptor, siendo la propuesta actual darle el nombre de Defectos Hereditarios en el Receptor de vitamina D (7).
El tipo I, también conocido como forma pseudo-deficiente de vitamina D o raquitismo pseudovitamin D resistente, presenta defectuosa 1 alfa hidroxilación de la 25 hidroxivitamina D, impidiéndose la formación renal de la forma activa de la vitamina D. Es un trastorno autosómico recesivo reportado principalmente en Japón, Franco-Canadienses y más recientemente en Estados Unidos, Korea, Argentina, Dinamarca y Morocco, poblaciones en las cuales ha sido posible identificar a la fecha diversas mutaciones inactivantes en el gen localizado en el cromosoma 12 que codifica la 1 alfa hidroxilasa, también conocida como 25-hidroxivitamina D3 1 alfahidroxilasa ó CYP27B1 (8-11). En estos pacientes los niveles de 1,25(OH)2vitamina D3 se encuentran bajos, y los de 25 (OH) hidroxivitamina D3 son normales a altos. En su tratamiento la administración de vitamina D3 (colecalciferol), vitamina D2 (ergocalciferol), calcidiol (25 (OH) D2 o D3) no tienen utilidad terapéutica, puesto que deben ser finalmente dihidroxilados por la 1 alfa hidroxilasa para ser activos. El calcitriol y la 1alfa(OH)vitamina D3 en dosis fisiológicas se propone como la mejor alternativa terapéutica, pero el problema radica en establecer cuál es esa dosis, la que al parecer es muy individual.
La gran mayoría de los artículos que se encuentran en la literatura se refieren a formas familiares de déficit de 1 alfa hidroxilasa, las cuales se identifican desde la niñez, cuando se manifiestan con hipotonía, debilidad, falla para crecer, convulsiones por hipocalcemia y signos radiológicos típicos de raquitismo. La 1 alfa hidroxilasa es una enzima mitocondrial de la familia de la citocromo P450 (12), se detecta principalmente en los riñones, pero recientemente se ha establecido que puede estar presente en otros tejidos corporales como en los keratinocitos epidérmicos, macrófagos, colon, próstata y seno (13). Factores que regulan la actividad de esta enzima son la PTH, calcio, fósforo y 1,25(OH)2D.
En nuestro paciente el diagnóstico se practicó en forma tardía, y sin presentarse las manifestaciones descritas en niños; no tenemos una explicación concluyente al respecto, pero proponemos un déficit parcial de la enzima, o un defecto renal adquirido asociado a su patología de nefrolitiasis múltiple por ácido úrico. Es importante resaltar que en todos los casos presentados en la literatura universal siempre los criterios exigidos para su diagnóstico son la presencia de hallazgos de laboratorio idénticos a los reportados por nosotros, procediéndose luego a analizar el DNA genómico de los pacientes y el gen de la 1 alfa hidroxilasa, datos finales que de practicarse en nuestro paciente podrían determinar el tipo de mutación que presenta.
Referencias
1. Holick M F. Vitamin D and health: evolution, biologic functions, and recommended dietary intakes for vitamin D. Clin Rev Bone Miner Metab 2009; 7: 2-19. [ Links ]
2. Terkeltaub, RA. Clinical practice. Gout. N Engl J Med 2003; 349: 1647-55. [ Links ]
3. Becker MA; Chohan S. We can make gout management more successful now. Curr Opin Rheumatol. 2008 Mar; 20: 167-72. [ Links ]
4. Becker, MA, Schumacher, HR Jr, Wortmann, RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med 2005; 353:2450-61. [ Links ]
5. Vogt, B. Urate oxidase (rasburicase) for treatment of severe tophaceous gout. Nephrol Dial Transplant 2005; 20: 431. [ Links ]
6. Fukumoto S, Namba N, Ozono K, Yamauchi M, Sugimoto T, Michigami T, Tanaka H, Inoue D, Minagawa M, Endo I, Matsumoto T. Causes and Differential Diagnosis of Hypocalcemia -Recommendation Proposed by Expert Panel Supported by Ministry of Health, Labour and Welfare, Japan. Endocrine Journal 2008; 55: 787-94. [ Links ]
7. Liberman U A. Vitamin D-resistant diseases. J Bone Miner Res 2007; 22: 105-7. [ Links ]
8. Labuda M, Morgan K, Glorieux F H. Mapping autosomal recessive vitamin D dependency type I to chromosome I2qI4 by linkage analysis. Am J Hum Genet 1990; 47: 28-36. [ Links ]
9. Kitanaka S, Takeyama K I, Murayama A, Sato T, Okumura K, Nogami M, Hasegawa A, Niimi H, Yanagisawa J, Tanaka T, Kato S K. Inactivating mutation in the 25-hydroxivitamin D3 1 alfa hydroxylase gene in patient with pseudovitamin D-deficiency rickets. N Engl J Med 1998; 338: 653-61. [ Links ]
10. Yoshida T, Monkawa T, Tenehouse H, Goodyer P, Shinki T, Suda T, Wakino S, Hayashi M, Saruta T. Two novel 1 alfa hydroxylase mutations in French- Canadians with vitamin D dependency rickets type I. Kidney Int 1998; 54: 1437-43. [ Links ]
11. Kim C J, Kaplan L E, Perwad F, Huang N, Sharma A, Choi Y. Vitamin D 1-Hydroxylase Gene Mutations in Patients with 1 alfa Hydroxylase Deficiency. J Clin Endocrinol Metab 2007; 92: 3177-82. [ Links ]
12. Negri A L, Fradinger E. Nuevos factores que intervienen en la regulación de la 1 alfa hidroxilasa renal de la vitamina D. Nefrología 2005; 25: 602-7. [ Links ]
13. Holick M F. Vitamin D deficiency. N Engl J Med 2007; 357: 266-81. [ Links ]

