



![Utilidad del [99m] Tc TRODAT-1 en el diagnóstico de la enfermedad de Parkinson](/img/es/prev.gif)






Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActa Medica Colombiana
versión impresa ISSN 0120-2448
Acta Med Colomb v.35 n.3 Bogotá jul./sep. 2010
Presentación de Casos
Restrictive cardiomyopathy as a presentation of amyloidosis
Iván Darío Rendón,(1) Andrés Mauricio Acevedo,(2) Andrés Felipe Buitrago,(3) Bernardo Lombo,(4) Mónica Jaramillo,(5) César Emilio Barrera(6) • Bogotá, D.C.
(1) Medicina Interna, Fellow de Cardiología, Universidad El Bosque;
(2) Estudiante de Medicina Universidad de los Andes;
(3) Médico Cardiólogo, Medicina Interna, Fundación Santa Fe de Bogotá;
(4) Médico Internista, Fundación Santa Fe de Bogotá;
(5) Médico Cardiólogo, Medicina Interna, Fundación Santa Fe de Bogotá;
(6) Médico Cardiólogo, Ecocardiografista, Fundación Santa Fe de Bogotá. Bogotá, D.C.
Correspondencia: Iván Darío Rendón Múnera. Calle 119 No. 9-33. Fundación Santa Fe de Bogotá. Servicio de Cardiología y Hemodinamia. Tel: (571) 603 03 03, Fax: (571) 619 63 17. Bogotá, D.C. (Colombia).
E-mail: ivancho020@gmail.com
Recibido: 30/IX/10 Aceptado: 02/VII/10
Resumen
La cardiopatía amiloidea es una rara entidad que se puede presentar dentro del contexto de la amiloidosis sistémica. Su presentación clínica tiende a ser tardía, cuando la función cardiaca se encuentra altamente comprometida. Actualmente han surgido herramientas no invasivas que facilitan su diagnóstico. El tratamiento depende del subtipo y en su mayoría comprende medidas de soporte a la falla cardiaca para proteger otros órganos, junto con quimioterapia oral y trasplante autólogo de medula ósea. Adicionalmente, nuevos factores pronósticos se han postulado para su manejo. Se presenta un paciente que ingresó por disnea de rápida progresión, y cuyo cuadro clínico es consistente con falla cardiaca congestiva. Los estudios paraclínicos reportaron hipertrofia concéntrica del ventrículo izquierdo y un patrón de infiltrados ecogénicos en el miocardio con extensa fibrosis. Se diagnóstica cardiomiopatía restrictiva secundaria a amiloidosis AL. A partir del caso, se describen las características de la enfermedad y los nuevos adelantos en su manejo (Acta Med Colomb 2010; 35: 126-131).
Palabras clave: amiloidosis, cardiopatía restrictiva, diagnóstico, tratamiento.
Abstract
Cardiac amyloidosis is a rare disease that may occur as a component of systemic amyloidosis. Its symptomatic phase usually presents when cardiac function is highly compromised. Recently, some non-invasive diagnostic tools have emerged improving diagnosis. Treatment for this disease depends on its subtype. In most cases, it consists on supportive measures to improve heart failure and organ function, in addition with oral chemotherapy and autologous stem-cell transplantation. Alongside, new prognostic factors have been introduced. As an example, a clinical case about a patient who develops rapid-progression dyspnea and congestive heart failure is described. The ecocardiographic report stated a left ventricular concentric hypertrophy and ecogenic infiltrate patterns associated with extensive fibrosis. This leaded to the diagnosis of cardiac amyloidosis secondary to systemic amyloidosis. Based on this case, characteristics and new advances in its management are presented (Acta Med Colomb 2010; 35: 126-131).
Keywords: amyloidosis, restrictive cardiopathy, diagnosis, management.
Presentación del caso
Hombre de 69 años con historia previa de hipotiroidismo y dislipidemia en manejo farmacológico quien acude al servicio de urgencias debido a cuadro clínico de 20 días de evolución de disnea de grandes esfuerzos que evolucionó rápidamente a medianos esfuerzos asociado a edema en miembros inferiores.
Al examen físico de ingreso no tolera el decúbito, con presión arterial 100/62 media: 82 mm Hg, frecuencia cardiaca 92 por minuto, frecuencia respiratoria 24 por minuto, saturación 91% al aire libre, presenta ingurgitación yugular a 45 grados. A nivel cardiopulmonar: punto de máximo impulso en quinto espacio intercostal con línea paraesternal izquierda, ruidos cardiacos rítmicos, no se auscultó ni S3, ni S4, crépitos bibasales, reflujo hepatoyugular positivo y edema grado II en miembros inferiores.
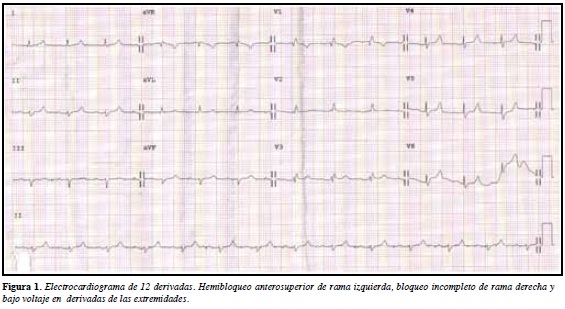
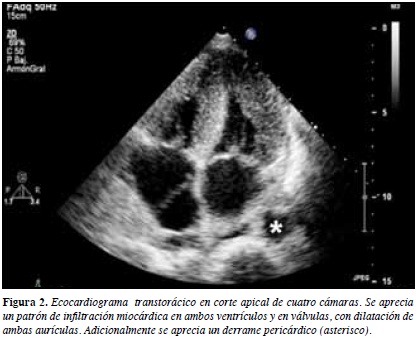
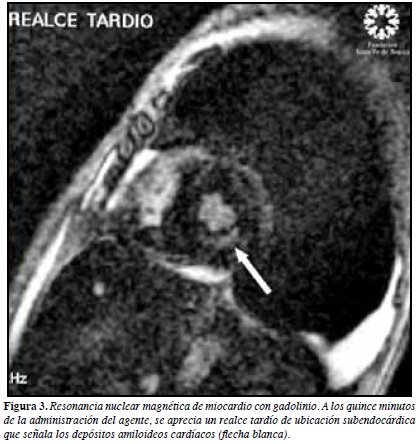
Electrocardiograma muestra hemibloqueo anterosuperior de rama izquierda, bloqueo incompleto de rama derecha y bajo voltaje en derivadas de las extremidades (Figura 1). Radiografía de tórax con derrame pleural bilateral. Ecocardiograma transtorácico muestra moderada hipertrofia concéntrica de ventrículo izquierdo (VI) compatible con posible infiltración amiloidea, disfunción diastólica restrictiva con fracción de eyección de 60% (Figura 2). Resonancia nuclear magnética reporta cardiomiopatía hipertrófica de características amiloideas (Figura 3), por lo que se realiza cateterismo cardiaco visualizándose hipertrofia del ventrículo izquierdo sin obstrucción al tracto de salida con fracción de eyección del 60%, se toma biopsia miocárdica reportándose amiloidosis con extensa fibrosis.
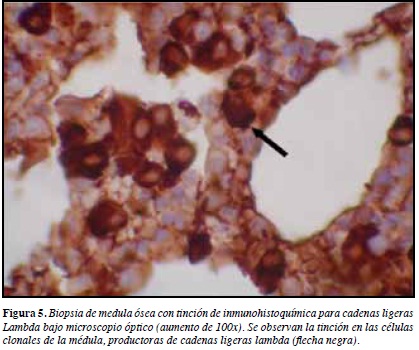
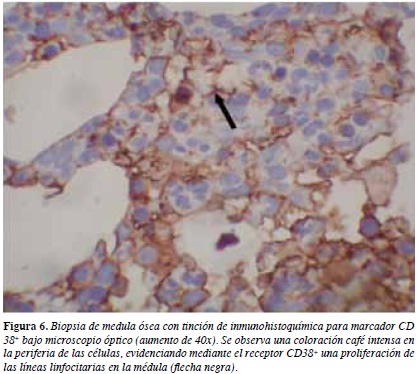
Biopsia de medula ósea reporta población plasmática 9% con cadenas ligeras lambda (Figuras 4, 5 y 6). Beta 2 microglobulina: 1779 ug/mL. Inmunoelectroforesis: banda monoclonal en gamma de 0.96 g/dL Ig G lambda.
Se diagnostica cardiomiopatía restrictiva secundaria a infiltración de proteína insoluble amiloidea tipo AL (IgG lambda), se da de alta a los 14 días del ingreso con manejo diurético para estabilización de falla cardiaca y posterior inicio de melfalán, prednisona y bortezomib como alternativa terapéutica.
Introducción
La amiloidosis describe la infiltración de los tejidos corporales por depósitos extracelulares de proteínas insolubles anormales originadas de un plegamiento incorrecto de proteínas normales, siendo estos resistentes a la proteólisis y generando al mismo tiempo un proceso inflamatorio local crónico. Aunque puede afectar un solo órgano, puede presentarse en forma sistémica. Por lo general los órganos más afectados comprenden el riñón, corazón, vasos sanguíneos, sistema nervioso e hígado. Se estima que la incidencia de este síndrome es de 10 por 1'000.000 casos/año (1). La amiloidosis cardiaca se define como la infiltración del tejido cardiaco por proteína amiloidea (de manera focal o dentro de un síndrome sistémico), y es un factor pronóstico determinante en los pacientes con amiloidosis sistémica, resultando en falla cardiaca congestiva, infarto y arritmias.
Clasificación
Por lo general la amiloidosis cardiaca se clasifica de acuerdo con la proteína precursora que conforma el depósito. Las más prevalentes se agrupan en cuatro grupos:
- Amiloidosis primaria (AL). En este subtipo la proteína amiloidea está compuesta por fragmentos monoclonales de cadenas livianas de inmunoglobulinas generadas por una discrasia de células B. En más del 80%, estas son gammapatías monoclonales benignas. Frecuentemente se presenta en la forma sistémica y hasta en un 90% de los casos involucra el corazón (1).
- Amiloidosis secundaria (AA). Se presenta asociada a enfermedades inflamatorias crónicas, como resultado de la acumulación de fibrillas de proteína A amiloide, la cual es un reactante de fase aguda. El órgano más afectado en este subtipo es el riñón y por tanto se presenta con proteinuria y falla renal. El compromiso del corazón en estos casos es raro y no tiene significancia clínica.
- Amiloidosis senil sistémica (SAA). En este subtipo, asociado a la edad, los depósitos se encuentran compuestos de la proteína transtiretina, sintetizada en el hígado y plexo coroideo. Se ha estimado la prevalencia en 25%-36% para mayores de 80 años (1). Presenta mayor prevalencia en hombres y en cuyos casos se ha visto mayor compromiso del tejido cardiaco. El deterioro clínico es lentamente progresivo asociada a síndrome del túnel del carpo, aunque en pocos casos la cardiomiopatía se presenta en forma severa.
- Amiloidosis hereditaria . Es un subtipo generado por una enfermedad autosómica dominante de mutaciones de las proteínas transtiretina, apolipoproteína I, entre otras. Por lo general, la proteína mutada más prevalente que involucra al corazón es la transtiretina asociadas a más de 80 sitios de mutación (2). Aunque es menos agresiva que la AL, puede generar falla cardiaca significativa, aunque tiende a asociarse más a neuropatía periférica.
Fisiopatología
El proceso degenerativo cardiaco se inicia a partir del depósito de proteínas insolubles en la matriz extracelular del tejido cardiaco. Los mecanismos que dirigen esta precipitación aún no se comprenden completamente, pero es determinante el tipo de anormalidad, lo que determina la producción de la proteína anómala. Por ejemplo, en pacientes con amiloidosis AL se ha observado un deterioro más rápido y un peor pronóstico, sugiriendo un efecto tóxico directo de las cadenas livianas de inmunoglobulinas (5). Estas proteínas penetran en forma de depósitos nodulares y filamentos ramificados que envuelven los miocitos, cuyo efecto neto es la disrupción de la estructura y función normal del tejido. La distribución de los depósitos no es uniforme y determina la presentación clínica: un patrón clásico difuso generará cardiomiopatía restrictiva y un patrón vascular causará obstrucción al flujo sanguíneo (5). En el primero, los depósitos inducen estrés oxidativo, reclutamiento de poblaciones celulares inflamatorias y remodelación de la matriz extracelular alterando el balance de metaloproteinasas y sus respectivos inhibidores y el sistema de conducción adyacente. El proceso inflamatorio recluta citoquinas y células citotóxicas que finaliza en necrosis de miocitos y depósitos de proteoglicanos, colágeno y otros componentes. En el segundo patrón, el depósito en la microvasculatura, en mayor proporción intramural, genera numerosos focos de isquemia y microinfartos, contribuyendo a la fibrosis (2). Como resultado final de lo anterior se produce un engrosamiento de la pared cardiaca de consistencia cauchosa y firme, empeorando la relajación, distensibilidad y función cardiaca. Esto se traduce en cardiomiopatía restrictiva identificable por presiones de llenado elevadas. Concomitantemente puede haber dilatación auricular por aumento de las presiones de llenado y su transmisión retrógrada.
Presentación clínica
Los signos y síntomas sistémicos son variados, sin embargo en el corazón se manifiestan usualmente como falla cardiaca diastólica en etapas avanzadas. Éstos incluyen edema periférico, hepatomegalia, hipotensión ortostática, ascitis, distensión yugular, fibrilación auricular e insuficiencia valvular. La afectación del tejido de conducción se relaciona con mayor frecuencia en la forma hereditaria, mientras que el deterioro diastólico prevalece en el subtipo AL (3). La isquemia miocárdica es rara, pero cuando se presenta como angina de pecho se debe alteraciones microvasculares imperceptibles a la angiografía coronaria (3). Pueden ocurrir síncopes por neuropatía autonómica o arritmias cardiacas con pobre reserva cardiaca.
Un cuarto de los pacientes con amiloidosis AL demuestran neuropatía autonómica (3). La severidad del cuadro se correlaciona con grados variables de infiltración miocárdica y la composición del depósito según el subtipo: la amiloidosis primaria presenta depósitos extensos y se manifiesta con disfunción cardiaca significativa. Los depósitos en otros subtipos por lo general se dan en focos aislados y con un compromiso más leve (4).
Diagnóstico
Métodos no invasivos
Ecografía: puede hallar características sugestivas de la enfermedad. Sin embargo, el diagnóstico debe realizarse en correlación con la presentación clínica y otros exámenes. Es imposible distinguir entre los diversos tipos de infiltrados amiloideos sólo haciendo uso de la ecografía. El signo ecográfico más comúnmente encontrado es el engrosamiento del ventrículo izquierdo, en ausencia de hipertensión. Sin embargo su especificidad es baja por otras patologías hipertrofiantes que también engrosan el miocardio. Una mejor rendimiento diagnóstico se obtiene al combinar el engrosamiento miocárdico con electrocardiograma (ECG) de bajo voltaje, con reportes de sensibilidad de 72%-79% y especificidad de 91%-100% (1). Otro signo ecográfico lo constituye la ecogenicidad del miocardio con "centelleos granulares" descritos por Siqueira-Filho como patognomónicos de amiloidosis cardiaca. Sin embargo, la sensibilidad de este hallazgo no es tan buena debido a que no siempre se hallan a la ecografía. Una combinación de ecocardiograma mostrando engrosamiento del ventrículo izquierdo, dilatación biauricular, valvas engrosadas y derrame pericárdico en un contexto de bajo voltaje en el ECG es altamente sugestivo de amiloidosis cardiaca (1).
En los casos avanzados, la ecografía Doppler es de alto valor para monitorear la progresión de la enfermedad. Permite evaluar el compromiso hemodinámico y funcional cardiaco. Los depósitos amiloideos limitan la relajación isovolumétrica, resultando en bajas velocidades de flujo de la diástole temprana a través de la válvula mitral, y aumento de las velocidades de llenado tardío diastólico por una mayor dependencia de la contracción auricular.
Electrocardiografía
Como una medida poco costosa, el ECG provee algunas claves diagnósticas, como bajo voltaje generalizado y patrones de pseudoinfarto. Murtagh y cols. encontraron en ECGs de 127 pacientes con amiloidosis AL que el 46% de los pacientes presentaron bajo voltaje (QRS de amplitud <10 mm en las derivaciones precordiales o <5 mm en las derivaciones de las extremidades) y 47% patrones de pseudoinfarto (infarto no evidente en la ecocardiografía). Se ha descrito que la característica de bajo voltaje y alta masa cardiaca puede ayudar en el diagnóstico. Un estudio encontró que en los pacientes con bajo voltaje y un grosor del tabique interventricular de >1.98 cm en la ecografía, presentaba un rendimiento diagnóstico con sensibilidad de 72% y especificidad del 91% (8). Aunque la validez externa del estudio se encuentra discutida, se ha planteado como criterio útil.
Son numerosas las arritmias cardiacas, pero la más frecuente es la fibrilación auricular asociada a la existencia de hipertrofia ventricular izquierda.
Resonancia magnética cardiaca
Esta técnica permite una visualización en tres dimensiones del miocardio, la evaluación de las cavidades, las paredes y su movimiento. La técnica de realce tardío con gadolinio provee una alta resolución y reproductibilidad. Se observa un mayor acortamiento del tiempo de relajación longitudinal subendocárdico (T1) o también un patrón típico de realce tardío subendocárdico difuso con gadolinio, lo que refleja la mayor infiltración amiloidea en el subendocardio con la expansión del espacio extracelular.
Gammagrafía nuclear
La administración de componente P amiloideo sérico radiomarcado, el cual se concentra en los depósitos amiloideos por unión dependiente de calcio a las fibras amiloideas, provee información única de la distribución y la extensión de los depósitos en todo el organismo, que además son útiles para el monitoreo del progreso y la respuesta al tratamiento (1).
Marcadores bioquímicos
Generalmente se hallan elevados marcadores séricos de daño cardiaco o estrés en la amiloidosis cardiaca. Las troponinas cardiacas específicas pueden elevarse como resultado de necrosis de miocitos e isquemia de pequeños vasos intramurales (2). La elevación de las presiones de llenado a su vez incrementan los niveles de péptido natriurético cerebral (BNP). Dispenzieri y cols postulan que estos marcadores séricos, en especial la medición del NT-proBNP (precursor del BNP) asociado a las troponinas cardiacas, permiten valorar el grado de disfunción cardiaca y una estratificación de riesgo exacta (6).
En etapas asintomáticas de pacientes con amiloidosis, un valor normal de BNP indicaba una probabilidad de amiloidosis cardiaca cercana a 0% (7). Se postula también su papel pronóstico evaluado en pacientes con quimioterapia en quienes se reducen los niveles de NT-proBNP con una mejor supervivencia (1, 5, 6). No se ha definido la utilidad del tamizaje con mediciones de NT-proBNP y se requiere evidencia adicional (1). Concordante con el efecto directo tóxico de las cadenas livianas de inmunoglobulinas, la medición de cadenas livianas libres (CLL) séricas se correlacionan con los niveles de NT-proBNP y la supervivencia. Se propone su utilidad como factor diagnóstico con una sensibilidad del 85%-98% (1).
Algunos estudios han mostrado que la reducción del 50% de los valores de CLL se asocia con mejoría clínica a pesar de no disminuir el grosor de la pared ventricular, y es un signo de buen pronóstico y adecuada respuesta al tratamiento con quimioterapia (1).
Métodos invasivos
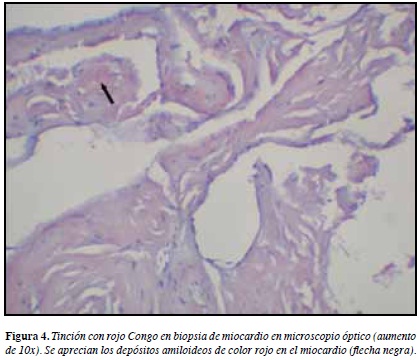
El diagnóstico definitivo se establece a través del análisis histológico del tejido cardiaco. La tinción del mismo con Rojo Congo identifica depósitos rosa amorfos en la microscopía de luz que bajo microscopía polarizada muestran birrefringencia verde.
Cuatro muestras endomiocárdicas aseguran casi un 100% de sensibilidad diagnóstica. Sin embargo, en forma menos invasiva se puede diagnosticar amiloidosis sistémica a través de observación de los depósitos en panículo adiposo, submucosa rectal o gingival. De ellas se prefiere la aspiración adiposa abdominal por mayor sensibilidad (84%-88%) y menores complicaciones (1). Hasta un 10% de pacientes amiloidosis AL no muestran anticuerpos monoclonales detectables, y es importante la biopsia de médula ósea para diagnosticar la presencia de poblaciones de células plasmáticas clonales responsables de la producción de cadenas ligeras amiloidogénicas (3).
Tratamiento
El manejo de la amiloidosis cardiaca tiene dos objetivos principales:
- Medidas de soporte para la mejoría de los síntomas y la sobrecarga de líquidos.
- Reducción de formación de fibras precursoras y depósitos amiloideos.
El manejo de soporte en la falla cardiaca se fundamenta en los diuréticos suministrados cuidadosamente con balance diario de fluidos y ajuste de dosis (1). Los b-bloqueadores se asocian a bradicardia e hipotensión (1). Los bloqueadores del canal de calcio y la digoxina se unen a las fibras amiloideas generando efectos tóxicos (2). Se debe establecer un monitoreo diario de peso e ingesta de sal. En los casos de trombos cardiacos, fibrilación auricular o falla de contracción auricular está indicada la anticoagulación (1). El beneficio de aplicar terapia antiarrítmica no se ha asociado a mejoría en la sobrevida y se requieren estudios (2).
El manejo del proceso amiloidogénico depende del tipo de amiloidosis. En el subtipo AA, el manejo principal es tratar el proceso inflamatorio subyacente. Esta terapia se asocia a veces a regresión de la disfunción orgánica y una sobrevida promedio de 24 1/2 meses (2). Nuevos agentes biológicos incluyen inhibidores del factor de necrosis tumoral y de interleucina-1. En el subtipo senil, el tratamiento en general es de soporte con una sobrevida promedio de 75 meses (2). El subtipo hereditario incluye el manejo sintomático y el trasplante hepático, fuente de la transtiretina. El pronóstico es bueno especialmente en pacientes jóvenes y la sobrevida promedio a cinco años es de 60%-77% (2).
El tratamiento de la amiloidosis AL se basa en quimioterapia hacia la población plasmática clonal. Los pacientes con amiloidosis avanzada toleran pobremente la quimioterapia y una proporción de clones son refractarios incluso a altas dosis terapéuticas. El esquema más efectivo hasta el momento parece ser quimioterapia a altas dosis con trasplante autólogo de médula ósea (7). Se ha observado con melfalán a altas dosis (140 mg/m2) altas tasas de respuesta hematológica (53% con remisión completa con inmunofijación negativa del 33%), asociado a tasas de mortalidad del 16% (6).
En la actualidad se encuentra en desarrollo el bortezomib (inhibidor del proteasoma) en fase de investigación I y II. El tratamiento con 1.6 mg/m2 semanal cada 35 días asociado a dexametasona mostró respuesta hematológica del 83% con una remisión completa en un 39% y un tiempo promedio de tratamiento de 0.9 meses (6).
Se ha evaluado el trasplante cardiaco como alternativa terapéutica, mostrando recaída del proceso amiloidogénico sobre el órgano trasplantado, sin que la quimioterapia disminuyera la mortalidad (1).
Conclusión
La cardiopatía amiloidea es una rara entidad que se puede presentar dentro del contexto de la amiloidosis sistémica. Su presentación clínica es variada y tiende a ser tardía, cuando la función cardiaca se encuentra comprometida, requiriendo alto nivel de sospecha.
La valoración ecocardiográfica y electrocardiográfica asociada a la presentación clínica permiten un diagnóstico no invasivo más exacto. El manejo incluye quimioterapia a altas dosis con trasplante de médula ósea vigilado a través de niveles de NT-proBNP y de cadenas livianas libres. Un diagnóstico temprano reduce la morbimortalidad en estos pacientes.
Agradecimientos
Los autores agradecen al Servicio de Cardiología y Hemodinamia del Hospital Universitario Fundación Santa Fe de Bogotá por su participación en el desarrollo del caso.
Referencias
1. Selvanayagam JB, Hawkins PN, Paul B, Myerson SG, Neubauer S. Evaluation and management of the cardiac amyloidosis. J Am Coll Cardiol 2007; 50: 2101-10. [ Links ]
2. Shah KB, Inoue Y, Mehra MR. Amyloidosis and the heart: a comprehensive review. Arch Intern Med 2006; 166: 1805-13. [ Links ]
3. Maredia N, Ray SG. Cardiac amyloidosis. Clin Med 2005; 5: 504-9. [ Links ]
4. Duque M, Marin JE, Luis EM, Uribe W, Velasquez JE. Cardiopamiopatía amiloidea. Rev Col Cardiol 2005; 11: 389-96. [ Links ]
5. Sawyer DB, Skinner M. Cardiac amyloidosis: shifting our impressions to hopeful. Curr Heart Fail Rep 2006; 3: 64-71. [ Links ]
6. Merlini G, Palladini G. Amyloidosis: is a cure possible?. Ann Oncol 2008; 19: 63-6. [ Links ]
7. Chau EM, Chow WH, Wang E, Kwong YL. Cardiac amyloidosis - experience in a tertiary cardiac referral centre. Int J Cardiol 2008; 124: 264-6. [ Links ]
8. Rahman JE, Helou EF, Gelzer-Bell R, et al. Noninvasive diagnosis of biopsyproven cardiac amyloidosis. J Am Coll Cardiol 2004; 43: 410 -5. [ Links ]

