










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.38 no.2 Bogotá Apr./June 2013
Revisión
Reacciones medicamentosas severas
Síndrome Stevens Johnson y síndrome DRESS
Severe drug reactions
Stevens Johnson syndrome and DRESS syndrome
Mauricio Torres, Edgar Olmos, Bogotá, D.C. (Colombia)
Dr. Mauricio Torres Pradilla: Dermatólogo Hospital de San José;
Dr. Edgar Olmos Olmos: Dermatólogo, Jefe Servicio de Dermatología, Hospital de San José. Bogotá, D.C. (Colombia).
Correspondencia. Dr. Mauricio Torres Pradilla, Hospital de San José. Bogotá, D.C. (Colombia).
E-mail: mtopra@gmail.com
Recibido: 06/III/2012 Aceptado: 19/III/2013
Resumen
Propósito: revisar y actualizar el síndrome de Steven-Johnson y el síndrome DRESS, que a pesar de no ser las reacciones medicamentosas más frecuentes, sí constituyen las más severas.
Fuente de datos: Realizamos una revisión sistemática de la literatura médica, expuesta en las bases de datos: Pubmed, Medline y Cochrane.
Selección de estudios: limitamos la búsqueda a artículos de revisión de literatura, guías de manejo y metaanálisis, publicados en inglés y español, sin límite en edad o sexo, utilizando términos MESH: Stevens-Johnson syndrome, DRESS syndrome. Los acoplamos a términos como assessment, associations, update y review.
Extracción de datos y resultados: realizamos una lectura inicial de 338 trabajos, encontrando que varios de las referencias eran citadas en otros trabajos, por lo que seleccionamos 10 artículos y con base en estos realizamos la lectura crítica, clasificación y ordenamiento, para luego proceder a la redacción del texto.
Conclusiones: hemos revisado dos importantes reacciones medicamentosas, entidades con un espectro clínico y etiopatogénico muy distinto, y numerosas asociaciones medicamentosas, sobre las que no existen realmente concensos globales de tratamiento, pero en la que todos los autores revisados coinciden en la suspensión de los medicamentos, como primera línea de tratamiento.
Palabras clave: síndrome Stevens-Johnson, síndrome DRESS, actualización, revisión, asociaciones, medicamentos, enfoque.
Abstract
Purpose: to review and update the Stevens-Johnson syndrome and DRESS syndrome, which despite not being the most frequent drug reactions, are the most severe ones.
Data sources: we conducted a systematic review of the medical literature, as described in the databases: PubMed, Medline and Cochrane.
Study selection: we limited the search to articles of literature review, management guidelines and meta-analysis published in English and Spanish, with no limit on age or sex, using MESH terms: Stevens-Johnson syndrome, DRESS syndrome. We linked them to terms such as assessment, associations, update and review.
Data extraction and results: we performed an initial reading of 338 studies, finding that several of the references were cited in other works, so we selected 10 articles and based on this we performed the critical reading, sorting and ordering, and then proceeded to the text drafting.
Conclusions: we reviewed two important drug reactions, clinical entities with very different clinical and etiopathogenic spectrum and numerous drug combinations on which there is not a global treatment concensus, but in which all authors agree in the drug suspension as the frst line of treatment.
Key words: Stevens Johnson syndrome, DRESS syndrome, update, review, association, medications, assessment.
Introducción
Las reacciones medicamentosas son un problema frecuente, no sólo por la gran diversidad de medicamentos existentes, sino por la variedad clínica de presentaciones de éstas y el desconocimiento por la mayoría de especialistas en su enfoque y manejo.
Es por esto que quisimos realizar la revisión de dos entidades, que a pesar de no ser tan frecuentes, sin ser raras o exóticas, pueden tener consecuencias catastróficas. Estas entidades clínicas corresponden al síndrome de Stevens-Johnson (SSJ), con sus variables de presentación clínica SSJ, SSJ/NET (síndrome de Stevens-Johnson/necrólisis epidermicotóxica) y NET (necrólisis epidermicotóxica), el cual consiste en una enfermedad ampollosa, por desprendimiento epidérmico y erosión, con compromiso de piel y mucosas, usualmente posterior a la exposición a un medicamento específico, principalmente carbamazepina, fenitoína y trimetropin-sulfametoxazol, entre otros. La otra entidad que vamos a revisar es SHDI (síndrome de hipersensibilidad a medicamentos inducida), o síndrome DRESS (por sus siglas en inglés, drug reaction with eosinphilia and sistemic symptoms, reacción medicamentosa con eosinofilia y compromiso sistémico), el cual consiste en una erupción cutánea (variable según el estadio de la enfermedad), acompañada de fiebre, leucocitosis con eosinofilia o linfocitosis atípica, adenopatías y compromiso de órganos internos, principalmente hígado y riñón. Ambas enfermedades tiene un pronóstico pobre, letal en su mayoría, si no son identificadas y tratadas.
Obtención de datos
Realizamos una revisión sistemática de la literatura medica, expuesta en las bases de datos: PubMed, Medline y Cochrane. Limitamos la búsqueda a artículos de revisión de literatura, guías de manejo y metaanálisis, publicados en inglés y español, sin límite en edad o sexo, utilizando términos MESH: Stevens-Johnson syndrome, DRESS syndrome, posteriormente acoplamos a términos como assessment, associations, update y review. Al final 338 trabajos, de los cuales al realizar una lectura inicial encontrando que varios de las referencias eran citadas en otros trabajos, ya fuera por sus resultados o fecha en que fueron escritos, por lo que seleccionamos al final 10 artículos, a los que realizamos lectura crítica, clasificación y ordenamiento, para luego proceder a la redacción del texto.
Resultados 1. Síndrome Stevens-Johnson
Definición
El síndrome de Stevens-Johnson (SSJ) y la necrólisis epidermicotóxica (NET) consisten en una enfermedad ampollosa severa frecuentemente causada por ingesta de medicamentos, caracterizado por desprendimiento epidérmico y erosión de las mucosas (1). Estas entidades consisten dos espectros de presentación de la misma entidad, diferenciadas entre sí por el grado de desprendimiento epidérmico, confiriendo mayor severidad a la NET . El pronóstico puede ser evaluado al aplicar el test de severidad de la enfermedad SCORTEN.
Historia
Descrito por primera vez en 1922, por los médicos americanos Stevens y Johnson, como síndrome mucocutáneo agudo, caracterizado por conjuntivitis purulenta severa, estomatitis severa con necrosis extensa en mucosas y lesiones cutáneas "eritema multiforme like", años después se denominaría síndrome de Stevens-Johnson, como una enfermedad mucocutánea severa, usualmente producida por medicamentos, con un curso prolongado y potencialmente letal, que debe ser diferenciada del eritema multiforme mayor.
En 1956, Alan Lyell, describió cuatro casos, de lesiones severas que denomino necrólisis epidermicotóxica, a partir de ese momento se describieron muchos casos clínicos adicionales, pudiéndose establecer la asociación con medicamentos.
Actualmente se consideran como dos espectros de presentación clínica de una misma entidad (2, 3).
Epidemiología
Es una enfermedad rara, incidencia global de dos casos por millón de habitantes, con un impacto social significativo debido una tasa de mortalidad de 20-25%, incapacidad prolongada y rechazo por parte de los sobrevivientes al uso de medicamentos (1).
Etiología y patogénesis
La patogénesis del SSJ/NET no es del todo entendida en el momento, evidencia reciente muestra que consiste en una entidad multifactorial, en adición a la predisposición genética se han implicado diversos mecanismos inmunes (4).
La susceptibilidad genética ha mostrado tener un papel muy importante en la patogénesis, debido a casos familiares y reacciones poblacionales a medicamentos específicos. En el momento se ha identificado el antígeno leucocitario humano (HLA)-B*1502 como altamente asociado a SSJ inducido por carbamazepina en la población China, estos hallazgos no han podido ser identificados en la población blanca (5). Otro estudio encontró alta asociación de HLA-B*5801 con SSJ y TEn inducido por alopurinol en japoneses, igualmente estos hallazgos no han sido extrapolados a la población blanca (5).
Los hallazgos histopatológicos principales en SSJ y NET consisten en queratinocitos apoptóticos difusos, este proceso no ha sido claramente elucidado, pero se ha sugerido su origen por tres vías: interacción Fas/Fas ligando, los cuales se encuentran elevados en los queratinocitos y estarían induciendo las caspasas, y por lo tanto la apoptosis celular; linfocitos T citotóxicos y linfocitos T natural Killer (nK) con la producción de perforinas y granulisina; por último el factor de necrosis tumoral alfa (TNF-a), el cual se encuentra elevado, induciendo la sobreproducción de complejos mayor de histocompatibilidad (MHC) clase I, haciendo más susceptibles a los queratinocitos a los linfocitos T citotóxicos (4).
Clínica
Fase aguda. Los síntomas iniciales son inespecíficos e incluyen fiebre, ojo lloroso y disfagia. Estos síntomas se presentan días antes del inicio de las manifestaciones cutáneas. Los sitios de presentación cutánea inicial son piel de región preesternal, cara, palmas y plantas. Eritema y erosiones en mucosa bucal, genital y ocular están presentes en casi todos los pacientes, algunas veces están comprometidas también las mucosas respiratorias y gastrointestinales.
La morfología de las lesiones cutáneas es variada, iniciando como máculas eritematosas con tendencia a confuir o máculas eritemato-violáceas dolorosas (marcador de severidad), que rápidamente evolucionan a ampollas epidérmicas, signo de nikolsky + (Figuras 1 y 2).
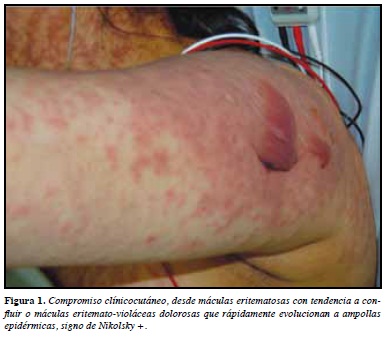
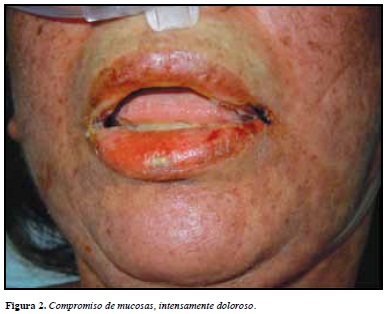
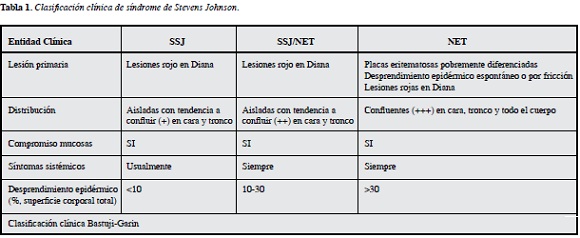
La extensión de las lesiones es el principal factor pronóstico, debido a que permite clasificar la enfermedad en SSJ (menos del 10%), NET (mayor del 30%) y la sobreposición de las dos entidades cuando el desprendimiento está entre 10 y 30% (Tabla 1).
Fase tardía y secuelas. Las secuelas son frecuentes, se presentan en 75% de los pacientes, consisten en hiper o hipopigmentación, distrofias ungueales, queratinización de la conjuntiva y queratoconjuntivitis sicca. Otros hallazgos incluyen lesiones de córnea y simblefaron.
Diagnóstico diferencial
Los principales diagnósticos diferenciales son las enfermedades autoinmunes, incluyendo dermatosis IgA linear, penfgoide ampolloso, y el síndrome de piel escaldada estafilocócica, este último con histopatología completamente diferente favoreciendo la diferenciación.
Diagnóstico
El diagnóstico de SSJ y NET es principalmente basado en los hallazgos clínicos; sin embargo, estos deben ser confirmados por biopsia y estudio histopatológico, enviando dos muestras para estudio inmediato por congelación (resultados en una hora), presentando queratinocitos apoptóticos y desprendimiento epidérmico, los hallazgos en coloración básica dependen del estadio de la lesión, temprano queratinocitos necróticos, y tardío necrosis epidérmica con ampollas subepidérmica.
Los exámenes de laboratorio son importantes para definir estado del paciente y realizar el test de SCORTEn el cual me permite determinar el pronóstico del paciente, por lo que se debe realizar hemograma completo, enzimas hepáticas, pruebas de función renal, glicemia, radiografía de tórax (6).
Medicamentos asociados
Más de 100 medicamentos han sido asociados a SSJ y NET desde su descripción, en el momento las principales recomendaciones de alerta existen sobre los siguientes medicamentos:
- Nevirapina
- Lamotrigina
- Carbamazepina
- Fenitoína
- Clotrimazol y otras sulfonamidas
- Sulfazalazina
- Alopurinol
- Oxicam - AINES
- Trimetropin-sulfametoxazol
- Cefalosporinas
Tratamiento
El tratamiento de esta entidad consiste en dos pilares funda mentales, suspensión temprana del medicamento causal, entre más rápido sea suspendido el medicamento mejor el pronóstico del paciente, y el inicio de medidas de soporte intensivas, idealmente en unidad de cuidados intensivos o en unidad de cuidado de quemados (6).
El manejo de las medidas de soporte debe ser realizado de forma multidisciplinaria, con profesional especializado, buscando proteger la dermis y mucosas expuestas, detección temprana y tratamiento de infecciones, soporte nutricional, monitoreo estricto de líquidos y electrolitos, y manejo de analgesia.
Las medidas farmacológicas actualmente son contro-versiales, ninguna con indicación específica para ser usada, se pueden dividir en dos grupos, según el efecto deseado:
a) Terapia inmunosupresora: incluyendo ciclosporina A y corticosteroides sistémicos.
La ciclosporina A, asociado con rápida reepitelización y menor mortalidad, cuando se compara con ciclofos-famida y corticosteroides, utilizado a dosis de 3 mg/kg/ día, durante 8-24 días (6).
Los corticosteroides sistémicos, tienen propiedades antiinfamatorias, inmunosupresoras y antiapoptóticas por lo que podría ser útil en el tratamiento; sin embargo, su uso en SSJ y NET es controversial. Clásicamente se ha utilizado dexametasona IV 1.5 mg/kg/día, por tres días consecutivos, logrando según algunas revisiones disminución en el tiempo de reepitelización, pero comparativamente no modificando la tasa de mortalidad cuando se compara con medidas de soporte adecuadas (6).
b) Medidas antiapoptóticas: clásicamente utilizando inmunoglobulina humana (IVIg), debido a la presencia de autoanticuerpos contra FAS, y por lo tanto un potencial efecto antiapoptótico, con resultados variables en efectividad, la dosificación recomendada es de 1 g/kg/día durante 3-4 días. El estudio observacional europeo concluyó que ni los corticosteroides, ni la IVIg tiene efectos significativos sobre la mortalidad en comparación con medidas de soporte adecuadas (6). Otra opción terapéutica es la plasmaféresis, con reporte de resultados exitosos de hasta 80% en NET severo sin respuesta a ningún medicamento. El mecanismo de acción probable consiste la remoción de FAS Ligando.
La valoración temprana y manejo por oftalmología es indispensable, sobre todo para prevenir las secuelas, el tratamiento oftalmológico comprende lubricantes, antibióticos y esteroides tópicos, pacientes con compromiso de córnea severo pueden requerir trasplante.
Pronóstico
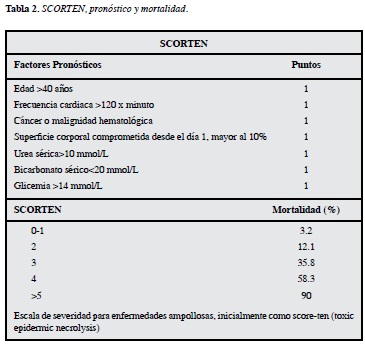
El síndrome de Stevens-Johnson y la necrólisis epider-micotóxica son enfermedades severas y potencialmente mortales. La mortalidad promedio en SSJ es de 1-5% y en NET es de 25-35%. Buscando evaluar el riesgo y pronóstico de los pacientes con SSJ y NET se han utilizado múltiples scores siendo el más aceptado en el momento el SCORTEN, el cual evalúa diversos parámetros como: edad, presencia de malignidad, taquicardia, desprendimiento epidérmico inicial, urea sérica, glicemia y bicarbonato (Tabla 2).
2. Síndrome de hipersensibilidad inducida (SHDI)/reacción medicamentosa con eosinofilia y síntomas sistémicos (síndrome DRESS)
Definición
El síndrome de hipersensibilidad inducido por medicamentos consiste en una reacción adversa potencialmente letal, caracterizada por rash, fiebre, leucocitosis con eo-sinofilia o linfocitosis atípica, adenopatías y compromiso órganos internos (riñón o hígado) (7).
Historia
No ha existido un término consistente para este síndrome, han sido usado varios términos para referirse, asociados principalmente al agente etiológico o la consecuencia patofisiológica, por ejemplo el síndrome de fenitoína, síndrome de hipersensibilidad por alopurinol, síndrome de dapsona, neumonía eosinofílica y dermatitis exfoliativa. Bocquet y colaboradores, en 1996, plantean el término rash medicamentoso con eosinofilia y síntomas sistémicos (DRESS), buscando simplificar la nomenclatura de estos síndromes (8). Descamps y colegas (1997), Hashimoto y colaboradores (1998) y Shiohara y colaboradores (1998) demuestran la relación entre esta reacción medicamentosa y reactivación de infección por herpes virus 6 (HHV-6), subsecuentemente este último grupo de investigadores decide acunar el término de síndrome de hipersensibilidad inducida por drogas (SHDi) publicándolo en 2003 (9). A pesar de compartir un mismo nombre, cada uno de los agentes etiológicos tiene unas características clínicas y patológicas específicas.
Epidemiología
La incidencia estimada es variable, siendo desde uno en 1000 hasta uno en 10000, en personas expuestas a los medicamentos asociados. La mortalidad es aproximadamente de 10% y está asociada principalmente al compromiso de órganos, siendo las más frecuentes insuficiencia hepática, falla renal y neumonitis intersticial (7).
Etiología
Este síndrome está asociado a unos tipos específicos de medicamentos (Tabla 3), algunos sin ninguna relación farmacológica o estructural entre sí, motivo por el cual la patogenia de esta entidad no es claramente entendida.

Varios factores podrían contribuir a su desarrollo, entre los que se han descrito son disminución de los niveles séricos de inmunoglobulinas, hipogammaglobulinemia transitorias y disminución de células T y B (8).
Clínica
La presentación clínica de esta entidad incluye lesiones cutáneas, variables consistentes en eritema, infltrados infamatorios y descamación en las lesiones crónicas, diseminados en todo el cuerpo y acompañado de edema facial, algunas veces queilitis, eritema faríngeo y pequeñas erosiones en mucosa oral. Adicionalmente se presenta con adenopatías y compromiso de hígado, riñón, pulmones o corazón (10). (Figuras 3 y 4).
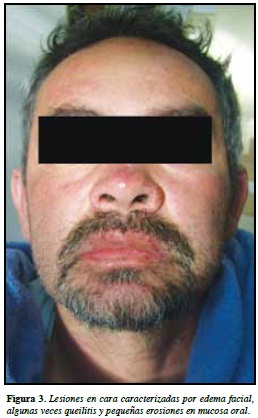
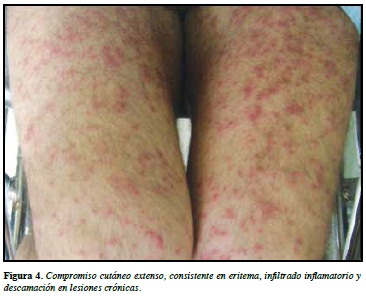
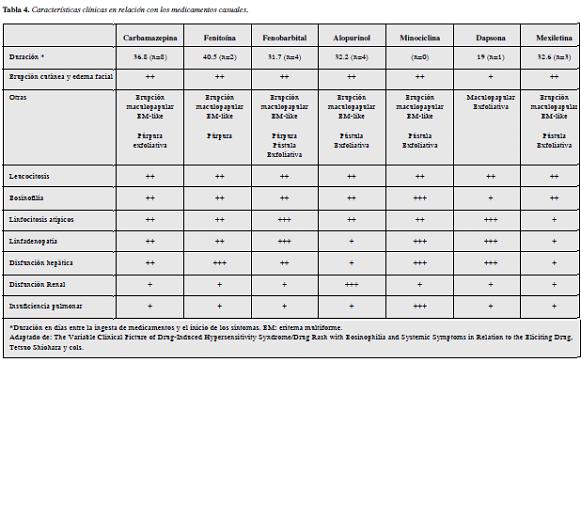
Todas estas manifestaciones son variables según el ti em po de evolución de la enfermedad y el tipo de medicamento asociado, según el análisis presentado por el grupo del Dr. Shiohara y colaboradores (7) (Tabla 4).
Otros compromisos sistémicos descritos son:
- Colitis/sangrado intestinal
- Diabetes mellitus
- Encefalitis/meningitis aséptica
- Hepatitis
- Nefritis intersticial
- Neumonitis intersticial/síndrome de dificultad respiratoria
- Miocarditis
- Serositis
- Síndrome de secreción inadecuada de hormona antidiurética
- Tiroiditis
Diagnóstico diferencial
Durante el primer episodio se puede confundir con el exantema maculopapular inducido por medicamentos, si se presentan pústulas se puede sospechas PEAG. Dependiendo el grado de infiltración de las lesiones se debe descartar linfomas o pseudolinfomas, lupus eritematoso inducido por medicamentos, enfermedad de Kawasaki.
Otros diagnósticos por tener en cuenta son infecciones agudas virales (sarampión), especialmente las que comprometen el hígado (virus Epstein-Barr, citomegalovirus, hepatitis A, hepatitis B e influenza) (10).
Diagnóstico
En el diagnóstico se han propuesto unos criterios especí-fcos para síndrome de DRESS por el Dr. Bocquet y colegas, en los que se incluye:
- Erupción cutánea medicamentosa.
- Anomalíashematológicas, eosinofilia(>1.5x109 eosinóflos/L) o la presencia de linfocitos atípicos.
- Compromiso sistémico, incluyendo adenopatías >2cm, hepatitis (elevación de transaminasas, dos veces del valor de referencia), nefritis intersticial, neumonía intersticial o carditis. Para el diagnóstico de síndrome de hipersensibilidad inducida por medicamentos, los grupos de investigación japoneses, proponen los siguientes criterios:
- Rash maculopapular, desarrollado posterior a tres semanas de iniciados los medicamentos específicos.
- Prolongación de síntomas dos semanas después de suspendido los medicamentos.
- Fiebre, >38°C.
- Anomalías hepáticas (ALT >100U/L).
- Anomalías en leucocitos, incluyendo: leucocitosis (>11x109 leucocitos/L), linfocitos atípicos (>5%) o eosinofilia (>1.5x109 eosinóflos/L).
- Linfadenopatía.
- 7.Reactivación por HHV-6.
Estudios complementarios como biopsia y estudio histopatológico, laboratorios clínicos e imagenológicos, permiten evaluar la enfermedad y ayudar en el diagnóstico diferencial, debido a que ninguno es el gold estándar para el diagnóstico.
Tratamiento
Los corticoides tópicos y sistémicos son la primera línea de tratamiento, aunque el compromiso sistémico es quien va a dirigir el enfoque terapéutico. Corticoides sistémicos iniciando con dosis moderadas y posterior disminución lenta, acompañado la suspensión del medicamento causal (10).
Conclusiones
Hemos revisado éstas dos importantes enfermedades, muy distintas entres sí en cuanto a etiología, fisiopatología, clínica e histopatología, siendo importante resaltar que las dos tienen en común la suspensión de los medicamentos, como primera línea de tratamiento, existen muchísimas discusiones sobre cuál es la forma más apropiada de manejarlas, pero algo en que la literatura es consistente es en que lo primero que debemos es suspender todos los medicamentos y realizar el soporte clínico necesario a nuestro paciente. Es por esto, que el médico general, a la vez que el especialista sea médico internista, dermatólogo, intensivista o cualquiera que sea la especialidad, reconozca estas entidades y realice el manejo oportuno, evitando así consecuencias catastróficas.
Agradecimientos
Agradecemos el apoyo dado por la biblioteca de la Fundación Universitaria de Ciencias de la Salud del Hospital de San José, al permitirnos acceso a todos los artículos de nuestra revisión a través de sus bases de datos electrónicas.
Conflictos de interés
Declaramos no haber recibido ningún tipo de financiación para esta revisión y no tener conflicto intereses.
Referencias
1. Mockenhaupt M, Viboud C, Dunant A, Naldi L, Halevy S, Bouwes Bavinck JN, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis: assessment of medication risks with emphasis on recently marketed drugs. The EuroSCAR-study. J Invest Dermatol 2008; 128(1): 35-44. [ Links ]
2. Harr T, French LE. Severe cutaneous adverse reactions: acute generalized exanthematous pustulosis, toxic epidermal necrolysis and Stevens-Johnson syndrome. Med Clin North Am 2010; 94(4): 727-42. [ Links ]
3. Auquier-Dunant A, Mockenhaupt M, Naldi L, Correia O, Schroder W, Rou-jeau JC. Correlations between clinical patterns and causes of erythema multiforme majus, Stevens-Johnson syndrome, and toxic epidermal necrolysis: results of an international prospective study. Arch Dermatol 2002; 138(8): 1019-24. [ Links ]
4. Koh MJ, Tay YK. An update on Stevens-Johnson syndrome and toxic epidermal necrolysis in children. Curr Opin Pediatr 2009; 21(4): 505-10. [ Links ]
5. Lonjou C, Borot N, Sekula P, Ledger N, Thomas L, Halevy S, et al. A European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to fve high-risk drugs. Pharmacogenet Genomics 2008; 18(2): 99-107. [ Links ]
6. Knowles S, Shear NH. Clinical risk management of Stevens-Johnson syndrome/ toxic epidermal necrolysis spectrum. Dermatologic therapy 2009; 22(5): 441-51. [ Links ]
7. Kano Y, Shiohara T. The variable clinical picture of drug-induced hypersensitivity syndrome/drug rash with eosinophilia and systemic symptoms in relation to the eliciting drug. Immunol Allergy Clin North Am 2009; 29(3): 481-501. [ Links ]
8. Cacoub P, Musette P, Descamps V, Meyer O, Speirs C, Finzi L, et al. The DRESS syndrome: a literature review. Am J Med 2011; 124(7): 588-97. [ Links ]
9. Shiohara T, Iijima M, Ikezawa Z, Hashimoto K. The diagnosis of a DRESS syndrome has been sufficiently established on the basis of typical clinical features and viral reactivations. Br J Dermatol 2007; 156(5): 1083-4. [ Links ]
10. Mockenhaupt M. Severe drug-induced skin reactions: clinical pattern, diagnostics and therapy. JDDG 2009; 7(2): 142-60. [ Links ]
