










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.39 no.2 Bogotá Apr./June 2014
Revisión
Síndrome hipereosinofílico y síndrome de Churg-Strauss ¿Espectro de una misma enfermedad? Nuevos conceptos
Hypereosinophilic syndrome and Churg-Strauss syndrome Spectrum of the same disease? New concepts
Elizabeth López-Piedrahíta1, Ruth H. Ramírez-Giraldo2, Carlos Jaime Velásquez-Franco4, Liliana Tamayo-Quijano3, Luis Fernando Pinto-Peñaranda4, Javier Márquez-Hernández4, Carlos Chinchilla-Mejía5, Ricardo Cardona-Villa6 • Medellín (Colombia)
1 Grupo de Alergología Clínica y Experimental; Medellín (Colombia)
2 Grupo de Alergología Clínica y Experimental. Pediatra-Alergóloga, IPS Universitaria. Docente Facultad de Medicina Posgrado de Alergología Clínica Universidad de Antioquia; Medellín (Colombia)
3 Grupo de Alergología Clínica y Experimental. Dermatóloga-Alergóloga, Clínica de la Universidad Pontificia Bolivariana; Medellín (Colombia)
4 Internistas- Reumatólogos, Hospital Pablo Tobón Uribe; Medellín (Colombia)
5 Grupo de Alergología Clínica y Experimental. Docente Facultad de Medicina Posgrado de Alergología Clínica Universidad de Antioquia. Pediatra-Alergólogo, Hospital Pablo Tobón Uribe; Medellín (Colombia)
6 Grupo de Alergología Clínica y Experimental. Docente Facultad de Medicina posgrado de Alergología Clínica Universidad de Antioquia. Alergólogo Clínico, Magister Inmunología, IPS Universitaria. Medellín (Colombia). Correspondencia. Dra. Elizabeth López. Medellín (Colombia).
E-mail: lopez55eliza@yahoo.es
Recibidio: 10/IX/2013 Aceptado: 12/V/2014
Resumen
Propósito de la revisión: revisar la literatura actual sobre el síndrome hipereosinofílico y establecercuáles son las diferencias y semejanzas con el síndrome de Churg-Strauss. Fuente de datos: revisión estructurada de la literatura médica, en las bases de datos Pubmed, Medline y Cochrane.
Selección de estudios: se limitó la búsqueda a los artículos de revisión, guías de manejo y meta-análisis, sin límites en idioma ni fecha de publicación, utilizando los términos MESH: hypereosinophilic síndrome, eosinophilic, Churg-Strauss sindrome. Se acoplaron a términos como: update, review, associations.
Extracción de datos: se hizo la lectura del resumen de 232 trabajos de los cuales se seleccionaron 111 que eran representativos para el tema de esta revisión, después de una clasificación y ordenamiento, se procedió a la redacción del texto.
Conclusiones: el síndrome hipereosinofílico comprende un grupo heterogéneo de enfermedades con presentaciones clínicas muy variadas, para algunos autores el síndrome de Churg-Strauss o más recientemente conocido como granulomatosis eosinofílica con poliangiítis hace parte de este gran síndrome pero para otros, deben considerarse como entidades aparte. (Acta Med Colomb 2014; 39: 174-184).
Palabras clave: síndrome hipereosinofílico, eosinofilia, síndrome de Churg-Strauss, granulomatosis eosinofílica con poliangiítis, actualización.
Abstract
Purpose of review: to review the current literature on the hypereosinophilic syndrome and establish which are the differences and similarities with Churg-Strauss syndrome.
Source of data: structured review of the medical literature in databases Pubmed, Medline and Cochrane.
Study selection: the search was limited to review articles, management guidelines and meta-analysis with no limits on language or publication date, using the MESH terms: hypereosinophilic syndrome, eosinophilic, Churg-Strauss syndrome. These were coupled to terms like:update, review, association.
Data extraction: reading of the summary of 232 studies was made. Of these, 111 that were representative for the subject of this review, were selected, and after an ordered classification system, the wording was performed.
Conclusions: The hypereosinophilic syndrome comprises a heterogeneous group of diseases with varied clinical presentations; for some authors, Churg-Strauss, more recently known as eosinophilic granulomatosis with polyangiitis, is part of this great syndrome, but for others these should be considered as separate entities. (Acta Med Colomb 2014; 39: 174-184).
Keywords: hypereosinophilic syndrome, eosinophilia, Churg-Strauss syndrome, eosinophilic granulomatosis with polyangiitis, update.
Introducción
El síndrome hipereosinofílico (SHE) es un trastorno que comprende un grupo heterogéneo de enfermedades caracterizadas por eosinofilia periférica, frecuentemente acompañadas de infiltración tisular, que resultan en una amplia variedad de manifestaciones clínicas que pueden ir desde la fatiga con lesiones cutáneas inespecíficas, hasta una fibrosis endomiocárdica, compromiso neurológico y comportarse como una enfermedad potencialmente mortal (1, 2). La gran variedad en las presentaciones clínicas y los diferentes órganos que en algún momento puede involucrar hacen difícil el diagnóstico del síndrome hipereosinofílico; por otro lado, para algunos autores el síndrome de Churg-Strauss (SCS) hace parte de las variantes del SHE, por lo que el objetivo de esta revisión se centra en establecer cuálesson las diferencias y semejanzas entre ambos, a la vez que se actualizan conceptos.
Obtención de datos
Se hizo una revisión sistemática de la literatura en las bases de: PubMed, Medline y Cochrane. Limitamos la búsqueda a artículos de revisión, guías de manejo y meta-análisis, sin límite en idioma o fecha de publicación, utilizando términos MESH: hypereosinophilic síndrome, churg strauss síndrome, eosinophilic, eosinophilic granulomatosiswith polyangiitis, posteriormente los acoplamos a términos como update, review, associations.
Se hizo la lectura inicial del resumen de 232 trabajos de los cuales se seleccionaron 111 que eran representativos para el tema de esta revisión, después de una clasificación y ordenamiento, se procedió a la redacción del texto.
Resultados
Criterios y clasificación
Hardy y Anderson, en 1968, fueron los primeros en referirse al SHE, al describir tres casos de pacientes con desórdenes leucoproliferativos con eosinofilia persistente (3), pero fue hasta 1975 cuando Chusid divulgó una seriede 14 pacientes y es el primero en proponer los criterios de diagnóstico, que incluyen: la eosinofilia en sangre periféricacon un recuento de más de 1500 células/µL durante más de seis meses consecutivos o muerte antes de este período, con signos y síntomas de SHE, ausencia de una causa obvia,por lo que agrega el término idiopático y daño o disfunción orgánica atribuible a los eosinófilos (4).
El Grupo de Trabajo de Enfermedades Hipereosinofílicas, en conjunto con la Sociedad Internacional del Eosinófilo realizó una publicación en el 2006,donde se discutieron las dificultades encontradas en los anteriores criterios y propusieron una clasificación sin el uso del término "idiopático", debido a que los avances tecnológicos han permitido identificar las causas de algunas de estas enfermedades y con el fin de capturar una amplia gama de trastornos que compartenmecanismos fisiopatológicos y clínicos (5) (clasificación en la Figura 1).
La problemática que surgió de los criterios clásicos ha sido que los pacientes con eosinofilia marcada y disfunción de tejido no deberían esperar seis meses para el diagnóstico, porque se retrasaría el tratamiento y generaría mayor infiltración tisular, que puede ser irreversible; por otro lado, un número definido de síndromes, incluidos el SCS, la mastocitosis sistémica, el angioedema episódico con eosinofilia (síndrome Gleich) han sido excluidos, a pesar de que sonclínicamente difícil de distinguir del SHE y son típicamente asociados a eosinofilia periférica. Además algunos pacientescon angioedema episódico con eosinofilia tienden a desarrollar una población de linfocitos aberrantes, sugiriendo que es una forma SHE variante linfocítica (6-9).
Otro punto controvertido es que los desórdenes eosinofílicos restringidos a un órgano tales como esofagitis, gastroenteritis, colitis eosinofílica (10, 11), neumonía eosinofílica crónica (12), celulitis eosinofílica (síndrome Wells)(13, 14) cumplen con la mayoría de los criterios de SHE, excepto la eosinofilia periférica, que sólo, en algunos casos superan las 1500 células/μL, a pesar de una clara relación del eosinófilo en la patogénesis de estas enfermedades tal como consideran algunos expertos (15). Además, la ausencia de manifestaciones clínicas en pacientes con eosinofilia mayor de 1500 eosinófilos puede reflejar una enfermedad en etapa temprana más que una anormalidad benigna del laboratorio (16).
En el artículo de Simon y colaboradores titulado "Refining the definition of hypereosinophilic síndrome" (17) proponen que la eosinofilia mayor o igual a 1500 debe presentarse en dos ocasiones y/o existir eosinofilia tisular sintomática además de la periférica, además de excluir las causas secundarias a infección, enfermedades alérgicas, eosinofilia inducida por medicamentos, hipoadrenalismo y neoplasias; los autores no definieroncon qué intervalo se deberían tomar las muestras ni definen eosinofilia marcada.
La OMS, estableció una clasificación semimolecular que a la fecha no ha sido actualizada, en la que incluyen los subtipos de la enfermedad eosinofílica como las neoplasias mieloides y linfoides con eosinofilia y anomalías genéticas, como las mutaciones de PDGFRA (de sus siglas en inglés: Platelet Derived Growth Factor Receptor Alpha), PDGFRB (Platelet Derived Growth Factor Receptor Beta), oFGFR1 (Fibroblast Growth Factor Receptor 1), la leucemiaeosinofílica crónica sin otra especificación (CEL, NOS), la hipereosinofilia variante linfocítica y el síndrome hipereosinofílico idiopático con los criterios clásicos de Chusid (18).
Nuevas propuestas de definiciones dehipereosinofilia y SHE
Durante las últimas décadas, varias clasificaciones de trastornos eosinofílicos han sido propuestas. Aunque los criterios y las definiciones se superponen, no se desarrolló un consenso mundial multidisciplinario, por lo que en 2011,en Austria, se organizó la Conferencia de Trabajo sobre los Trastornos y Síndromes de los Eosinófilos, con la participación de expertos en inmunología, alergia, hematología, patología y medicina molecular (19) en la que se definieron los nuevos criterios
Tradicionalmente, la eosinofilia ha sido dividida en leve (351-1500 Eos/uL), marcada o moderada (>1501Eos/uL) y masiva o grave (>5000Eos/uL) (19, 20), pero se debe teneren cuenta que en niños y en países en vía de desarrollo, ellímite de eosinófilos periféricos normales puede ser más laxo, como 500 o incluso 700 Eos/uL (21). En Colombia, la prevalencia de eosinofilias es de 11.63%, luego de un análisisde 153485 hemogramas realizados en el Laboratorio ClínicoHematológico de Medellín (22). En ese estudio se encontró que 94.51% de 18875 pacientes, tenían una eosinofilia leve (considerada como >351 Eos/uL), moderada en 5.12% y severa en 0.36%.
En el enfoque clínico, se debe tener en cuenta si la eosinofilia es clonal o no clonal; es decir, si hay o no compromiso de célula madre, y además si es transitoria, episódica o persistente (crónica) (23).
La propuesta por el panel de expertos es considerar la hipereosinofilia (HE) cuando existe una eosinofilia marcada ypersistente y/o se ha documentado eosinofilia tisular; en esteconsenso también se determinó que la eosinofilia en sangre periférica debería ser registrada en al menos dos ocasiones con un intervalo mínimo de cuatro semanas (excepto cuandola terapia inmediata es necesaria debido a la disfunción de órganos). No obstante, aclaran que no están disponibles datos de ensayos clínicos, por lo que se precisa de estudios adicionales para validar este punto, pero se diseñaron unos criterios para la HE y el SHE. (Tabla 1).
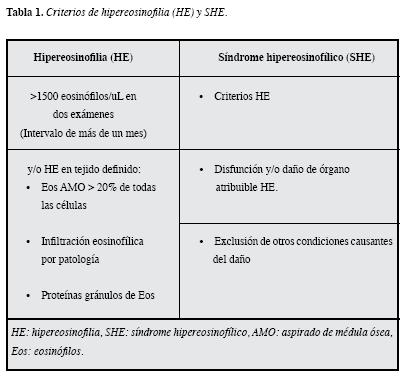
Con respecto a la HE tisular, consideran que debe aplicarse cuando se cumplan uno o más de los siguientes criterios: 1) El porcentaje de eosinófilos supera 20% de todas las células nucleadas en la biopsia medular (BM), 2) Que un patólogo determine que la infiltración de los tejidos por los eosinófilos es extensa cuando se compara con el rango fisiológico normal, 3) Determinación de gránulos eosinofílicos como la proteína básica mayor o la proteína catiónica del eosinófilo a través de un colorante específico y este tercer criterio es aplicable, incluso en ausencia de infiltración local masiva. Además aclaran que la HE tisular puede ocurrir en ausencia de HE en sangre, aunque en la mayoría de los casos también está presente, al menos una eosinofilia leve.Hasta la fecha sólo existe consenso para eosinofilia tisular en la esofagitis eosinofílica, que es mayor o igual a 15 Eos por campo de alto poder (CAP) o menos de 15 Eos si el paciente usa inhibidores de la bomba de protones pero hay evidencia de capas superficiales de Eos, microabscesos o gránulos extracelulares eosinofílicos (24). En otros tejidos serán necesarios estudios adicionales para tener medidas cuantitativas y parámetros objetivos avalados mundialmente.
El panel de expertos también sugirió una nueva clasificación para la HE, la cual dividieron en: HE primaria, neoplásica o clonal (HEN), HE reactiva o secundaria (HER),HE familiar (HEFA) y la HE de significado incierto (HEUS).
Resaltaron que estas variantes no representan un diagnóstico final, pero son puntos de partida para guiar el estudio en los pacientes, por ejemplo, un paciente clasificado como HEN puede tener una leucemia eosinofílica crónica (CEL) u otra neoplasia mieloproliferativa hasta que se confirme el tipo de mutación.
Las variantes HEFA y la HEUS son diagnósticos provisionales y necesitan estudios adicionales para excluir eldesarrollo de un SHE o de un desorden neoplásico.
El consenso estableció que el término SHE debe ser usado en cualquier variedad de HE con clara evidencia de daño orgánico por los eosinófilos, con la excepción de ciertas enfermedades, como la gastroenteritis eosinofílica, la neumonía eosinofílica, condiciones dermatológicas con eosinofilia y los síndromes que ya están bien establecidos, como síndrome de Gleich (angioedema cíclico con eosinofilia), SCS, síndrome de Omenn, síndrome de hiper-IgE y otras entidades en donde el compromiso del órgano es frecuentemente asociado con HE periférica pero el papel de la eosinofilia en la patogénesis no está bien comprendido.
En el año 2012 se publicó, además, un artículo de controversias sobre este mismo tema (25), donde reiteran que ladefinición de HE tisular necesita mayor precisión; y resaltannuevamente que los síndromes ya reconocidos no deberían agruparse con el término SHE.
Epidemiología
No existen datos confiables publicados sobre la incidencia y prevalencia del SHE; en los primeros estudios se estimaba que la prevalencia era de 14% (15). En Estados Unidos, la incidencia anual se estima en 1-2 casos por 200000 habitantes (26, 27). En la actualidad, series con grupos más grandes, encuentran que no es mayor de 10% (28).
Es poco frecuente en adultos y mucho más rara en niños, en los cuales están comúnmente asociadas anormalidades cromosómicas y rearreglos genéticos.
En general es más común en hombres que en mujeres, aunque entre sus variantes hay diferencias, para las formas mieloproliferativas, la relación hombre mujer es 9:1 y en las linfoproliferativas, indefinidas y familiares son de 1:1. Usualmente se diagnostica entre las edades de 20-50 años (2) pero se ha reportado un rango de diagnóstico entre los 7 y 77 años (29).
Fisiopatología
Los características más distinguibles del eosinófilo son: su núcleo multilobulado, y sus grandes gránulos eosinofílicos, rodeados por una doble membrana que contiene un core cristalino (30).
Sus funciones principales son: destruir parásitos invasores, fagocitar partículas bacterianas, producción de radicaleslibres y liberación de sus gránulos proteicos tóxicos que contienen cuatro proteínas denominadas: la proteína básica mayor (MBP), la proteína catiónica del eosinófilo (ECP), la neurotoxina derivada del eosinófilo (EDN) y la peroxidasa del eosinófilo (EPO); también son productores de más de tres docenas de citoquinas con funciones inmunorreguladoras y mediadores inflamatorios, como: el leucotrieno C4 (LTC4), el factor activador plaquetario (PAF) y el factor de crecimiento transformante β (TGF-β). Sin embargo, en ausencia de parásitos, los eosinófilos activados pueden causar destrucción e inflamación de los tejidos (31-33).
Diferentes estudios han descrito que la estructura del eosinófilo de pacientes con SHE es muy distinta a los de los sujetos sanos. Los eosinófilos de estos pacientes son más hipodensos, tienen alteraciones en el tamaño y forma, pierden el core granular, no tienen la doble capa lipídica en sus gránulos y éstos son más grandes y con mayor contenido de proteínas (34). Simon y cols (35) encontraron que los pacientes con HE expresan la cadena alfa del receptor de la IL-2 conocido como CD25 y estos eosinófilos CD25 positivos tienen la capacidad de liberar más proteínas y tienen mayor riesgo de de granulación, con el subsecuentedaño al tejido.
Por otro lado, algunos estudios en modelos animales y humanos (36, 37) han demostrado que no necesariamenteimporta el grado de infiltración eosinofílica encontrada, debido a que el contenido de los gránulos, al ser liberadoen los tejidos así sea de unos pocos eosinófilos, perturban la integridad de éste y generan los signos y síntomas del síndrome.
Manifestaciones clínicas
Existe una gran heterogeneidad clínica, desde una evolución asintomática hasta una enfermedad fatal. Los órganos afectados, en orden de frecuencia, son: piel, corazón, tracto gastrointestinal, sistema nervioso y pulmones (15, 38, 39). Los síntomas más reportados son: fatiga, prurito, tos, disnea, mialgias y fiebre (17, 40).La afectación cutánea generalmente se manifiesta con angioedema, urticaria, eritema, pápulas, nódulos, úlceras en mucosas, máculas pruriginosas o placas que semejan un eczema atópico. Además, también se han descrito, en menor proporción, púrpura palpable, livedo reticularis, eritrodermia, petequias, lesiones de eritema anular centrífugo, vasculitis, necrosis acral y hemorragias en astilla (2).
El daño cardiaco incluye tres etapas. La primera es la necrótica, que es asintomática, seguida por la fase trombóticay el estadio final, con fibrosis endomiocárdica y daño de las válvulas auriculoventriculares, que culmina en insuficienciacardiaca congestiva (41, 42).
En el tracto gastrointestinal se ha reportado infiltración eosinofílica, que en un inicio puede ser asintomática, o progresar hasta cuadros severos de abdomen agudo (10, 11, 43-45).
Las manifestaciones neurológicas pueden involucrar el sistema nervioso central y el periférico en donde pueden ocurrir: infarto cerebral o episodios de isquemia cerebraltransitoria secundarios a un trombo intravascular o intracardiaco. Los pacientes pueden experimentar encefalopatías, caracterizadas por cambios de comportamiento, confusión, ataxia y pérdida de la memoria. La neuropatía periférica representa el 50% de los síntomas neurológicos y se manifiestapor: déficit sensitivo asimétrico, parestesias, neuropatías motoras puras o déficit motor y sensitivo (46-48).
En los pulmones puede haber infiltración primaria de eosinófilos o daño secundario a la falla cardiaca congestiva o una embolia pulmonar. El derrame pleural fue la anormalidad más encontrada, pero la radiografía del tórax en la mayoría de los pacientes, es normal. Las sibilancias no son comunes y su hallazgo debe orientar a descartar un síndromede Churg-Strauss (49-52).
Variantes principales
La diversidad clínica del SHE y la ocasional transformación a una neoplasia, sugieren una heterogeneidad fisiopatológica. Como se describió previamente, algunos autores consideran que existen varios subtipos y los estudiosrecientes han identificado claramente dos trastornos hematológicos distintos con compromiso en la serie mieloide y linfoide, reconocidos como las variantes mieloproliferativa y linfocítica, respectivamente. Para Simon y colaboradores (16, 17) las variantes superpuesta, indefinida y asociada podrían hacer parte de la linfocítica.
SHE variante mieloproliferativa (SHE-M)
La variante mieloproliferativa se debe a una delección enel cromosoma 4, que genera la fusión de los genes FIP1L1 (Fip-1-like1) y PDGFRA, produciendo el gen de fusión F/Pque codifica para una proteína tirosina quinasa constitutiva que estimula la proliferación de eosinófilos y suprime, a su vez, su apoptosis (53-55).
La OMS ha considerado que cuando se identifique la mutación F/P se debe clasificar como leucemia eosinofílica crónica (18). Klion y colaboradores sugieren en el consensorealizado por el Grupo de Trabajo del Síndrome Hipereosinofílico en asociación con la Sociedad Internacional del Eosinófilo (5, 43) que los pacientes con ausencia detectable del gen de fusión F/P, mutación cromosómica relacionada, u otra evidencia de clonalidad eosinofílica, podrían considerarse que tienen SHE-M si cumplen cuatro de los criterios presuntivos (Tabla 2). Es importante resaltar que la demostración de clonalidad es difícil, especialmente en pacientes de sexo masculino.
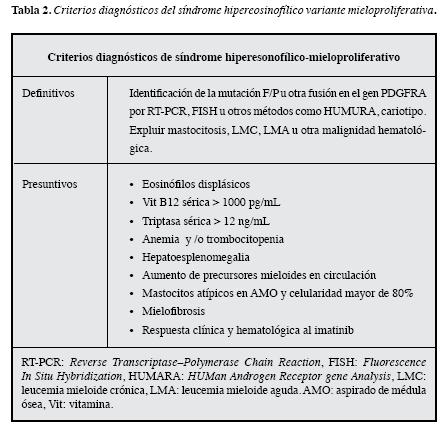
Entre otras características de esta variante, se han encontrado que puede simular una LMC u otros síndromes mieloproliferativos, lo que revela una forma más agresiva del SHE, con pronóstico desfavorable por la aparición de compromiso cardiaco, resistencia a los esteroides y por el riesgo a evolucionar hacia una neoplasia mieloide franca (56). Además, se han identificado que pueden existir otras anormalidades cromosómicas coexistentes como translocaciones, delecciones y trisomías 8, 15 y 21, especialmente enla población pediátrica (57, 58).
Aunque el compromiso cutáneo es más característico de la variante linfocítica, las hemorragias en astilla o la necrosis del lecho ungular deben alertar sobre la microembolia secundaria a compromiso endomiocárdico. Las úlceras en mucosas se asocian a un curso maligno y la muerte es comúnsi no hay tratamiento (59, 60).
El pulmón se afecta en más de 40% de los pacientes y la enfermedad se puede manifestar con edema pulmonar, derrame pleural, infiltrados intersticiales, imagen en vidrio esmerilado y pequeños nódulos (50, 61). La encefalopatía, lesión de neurona motora superior, polineuropatía o radiculopatía también se han reportado y la causa del daño es por lesión directa por las proteínas liberadas del eosinófilo (62).
SHE variante linfocítica (SHE-L)
Con la evidencia disponible hasta la fecha, la variante linfocítica obedece a una expansión clonal de linfocitos T(LT) aberrantes productores de citoquinas eosinofilopoyéticas con perfil Th2 como: son la IL-4, IL-5, IL-13, GM-CSF, las cuales desempeñan un papel regulador en la producción,diferenciación, reclutamiento, activación y sobrevida del eosinófilo, generando su incremento periférico y tisular (33, 63).
El inmunofenotipo más encontrado es el de LT CD3-CD4+ y en, menor proporción, se han identificado LT doblemente negativos (por ejemplo, los LT CD3+CD4-CD8-y LT CD3CD4+CD7-) y el LT CD3-CD4+CD5+, los cuales son LT inmaduros productores de citoquinas eosinofilopoyéticas con incapacidad para producir IFN-α (64, 65).
Las lesiones cutáneas son polimorfas, se observan en la mayoría de los pacientes y generalmente son la primera manifestación de la enfermedad (2, 37, 49); los antecedentesde atopia son frecuentes, la respuesta a esteroides es típica, al igual que la elevación de IgE, la quimoquina regulada porel timo y por activación conocida como TARC (T cellActivation-Regulated Chemokine) (28, 66) y la hipergammaglobulinemia policlonal por mayor nivel de IgG e IgM en suero, las cuales son características que ayudan a soportar el SHE-L. No obstante, el diagnóstico definitivo requiere la demostración de los LT aberrantes por citometría de flujo, lacual no siempre está disponible, por lo que el incremento decitoquinas eosinofilopoyéticas o el rearreglo del receptor dellinfocito T por PCR pueden ayudar a la identificación de estavariante sin ser criterios indispensables (67, 68) (Tabla 3).
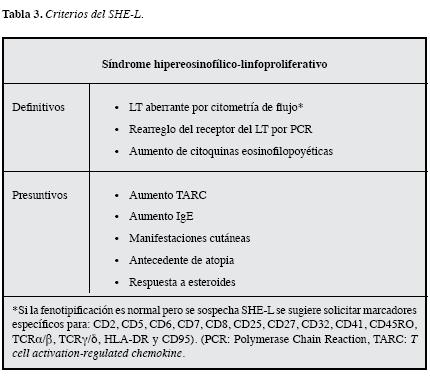
Es importante aclarar que, hasta la fecha, no hay consensopara los criterios del SHE-L y que los niveles séricos de IL-5 no son útiles como criterio aislado, ya que pueden estarnormales o elevados y no son útiles en evaluar respuesta terapéutica (69, 70).
Otras manifestaciones frecuentes en esta variante son las del tracto gastrointestinal como: dolor abdominal, náuseas, vómito, diarrea y disfagia, que se correlacionan con infiltración eosinofílica del sistema digestivo (44, 71, 72).
También están descritas artralgias, mialgias, artritis,tenosinovitis, adenopatías y fenómeno de Raynaud, (73, 74) ; por el contrario, es muy rara la fibrosis miocárdica, al igual que el compromiso pulmonar y neurológico (75-77).
Esta variante se considera que sigue un curso benignoy los clones de LT pueden permanecer estables por años,pero los pacientes deben ser observados porque, en casosmuy esporádicos, se comporta como una proliferaciónpremaligna y progresar a un linfoma, debido a que, con eltiempo, estos LT aberrantes acumulan anormalidades cromosómicas y adquieren nuevos marcadores de superficie,como CD25 y CD69. Se debe prestar especial atencióncuando se identifica el inmunofenotipo LT CD3-D4+, siel paciente persiste con HE severa a pesar de diferentestratamientos o desarrolla múltiples adenopatías (69, 78-80).
Semejanzas y diferencias entre el SCS y el SHE
Distinguir entre SCS y el SHE puede ser todo un reto,particularmente en las etapas tempranas de la enfermedad,debido a que ambos síndromes comparten característicasclínicas y serológicas; además, es mucho más difícil distinguirlos en pacientes con anticuerpos anticitoplasma deneutrófilos (ANCAs) negativos y ausencia histológica devasculitis (33, 81-83).
Desde el punto de vista fisiopatológico, que a la fechano está del todo dilucidado, el SCS, también conocidocomo granulomatosis eosinofílica con poliangiítis (83,89, 91, 111), se caracteriza por elevación de IgE, IL-5,eosinofilia periférica e infiltración tisular eosinofílica, loscuales son marcadores de respuesta Th2, como sucede enel SHE, principalmente en la variante linfocítica (SHE-L).Los eosinófilos generan activación del endotelio vascularal inducir la expresión de moléculas de adhesión ICAM-1y VCAM-1, llevando a una respuesta inflamatoria que espotenciada por las proteínas catiónicas de sus gránulos;adicionalmente la positividad de los ANCAs puede estarentre un 40-50% de los casos; este hecho podría sugerirun papel directo en el daño vascular y potenciarlo al activar neutrófilos y generar la liberación de enzimas líticas,radicales libres de oxígeno y activación del complemento(33, 84-86).
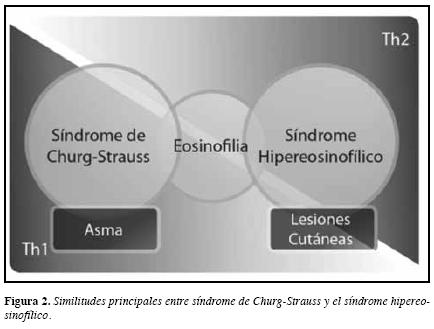
A medida que el SCS progresa, ya entrando en la fasevasculítica, algunos autores (87) consideran que va adquiriendo más un perfil Th1 o simplemente es una entidad quetiene varios subtipos, algunos de ellos indistinguibles delSHE (88) (Figura 2).
Al mismo tiempo, se considera que el SCS tiene tantoun componente alérgico como autoinmune, debido a quese ha identificado un mayor número circulante de LT, lapresencia de complejos inmunes y aumento sérico de laIgEy el factor reumatoide (89, 90). Sus criterios distintivos incluyen: vasculitis eosinofílica de pequeños vasos,la eosinofilia periférica mayor del 10% del diferencialde los leucocitos, asma generalmente de difícil control,infiltrados pulmonares, alteraciones en senos paranasales yneuropatía. La presencia de más de cuatro de estos criteriospropuestos por el Colegio Americano de Reumatología es sugestiva de SCS, pero, en ausencia de vasculitis confirmada, tienen un valor incierto (91, 92).
El diagnóstico del SCS generalmente se hace alrededorde los 50 años, pero se han reportado casos entre 30 y 66años de edad. Respecto a su presentación clínica, las manifestaciones cambian dependiendo del perfil de ANCAsque tenga el paciente; semejante a lo que ocurre en el SHE,en donde como ya se describió las manifestaciones sondiferentes dependiendo de su variante, pero en términosgenerales, el asma se manifiesta en 91.1%, la neuropatíaperiférica en 51.4%, compromiso de nariz, oídos y garganta 48%, lesiones cutáneas en 39.7%, infiltrados pulmonares ydaño cardiaco en 38.6% y 16.4%, respectivamente.
En los pacientes ANCAs positivos hay más frecuentemente manifestaciones en oídos, nariz y garganta,neuropatía periférica, daño renal y recaídas vasculíticas;mientras que en los ANCAs negativos, es más común lacardiomiopatía, compromiso abdominal, derrame pleural,dolor torácico y menor tasa de supervivencia (83).
Respecto a los paraclínicos ambos síndromes puedentener eosinofilia mayor a 1500 células, creatina fosfoquinasa (CPK) y troponinas aumentadas, eosinofilia en elaspirado de médula ósea; se ha encontrado que la proteínaC reactiva (PCR) y la velocidad de sedimentación globular(VSG) pueden estar más elevada en el SCS, lo mismo quela proteinuria y microhematuria.
A continuación se presentarán otras características clínicas y paraclínicas que comparten y en las que se diferencian ambos síndromes (Tabla 4).
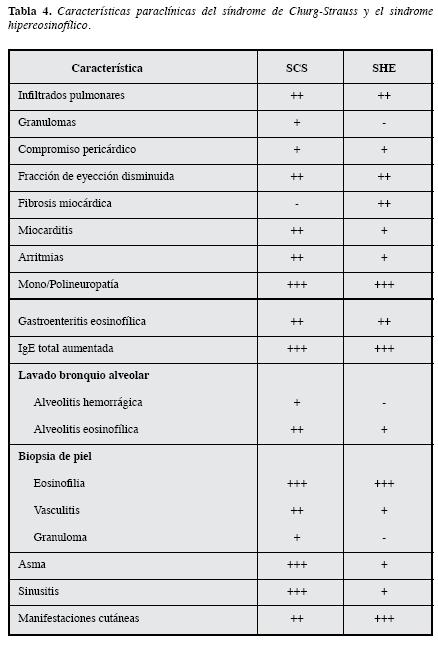
Diagnóstico
Los signos y síntomas del SHE, independiente de lavariante son comunes en muchos otros problemas desalud, lo que hace que el primer paso sea descartar otrasafecciones, principalmente las infecciones parasitarias yvirales, enfermedades alérgicas, neoplasias, enfermedadesdel sistema inmunológico, eosinofilas por medicamentos o químicos y el hipoadrenalismo, entre las más comunes(17, 18, 93).
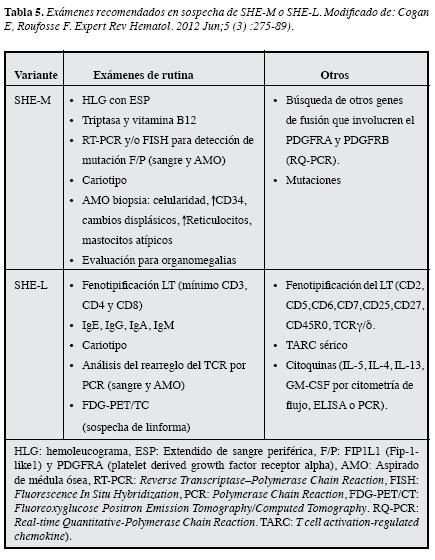
La evaluación se individualiza según la clínica pero,dentro de los protocolos que se tienen, se sugiere siempreincluir los que se esquematizan en la Figura 3 y Tabla 5.
Tratamiento
El objetivo principal del tratamiento es reducir los niveles de eosinófilos en sangre y tejidos, evitando de estamanera la progresión de la infiltración y el daño por laliberación de sus productos, por lo que la terapia a utilizarsedebe elegirse de acuerdo con la urgencia relativa que setenga en reducir estas células y poniendo en la balanza elriesgo/beneficio para el paciente antes de completar todoslos estudios necesarios para identificar la variante (94).
Los glucocorticoides, como prednisona, siguen siendola primera línea para la mayoría de los pacientes con SHE,excepto los que tienen la variante mieloproliferativa. Ladosis por utilizar varía desde 0.5-2 mg /Kg/día y se debeajustar según conteo de eosinófilos, signos y síntomas delpaciente. A pesar de la disminución de los eosinófilos ensangre periférica, la acción de estos medicamentos en elórgano infiltrado es variable de un paciente a otro y estámuy relacionada con el momento de inicio de la terapia (40,95). Se considera que la tasa de supervivencia es de másde 80% para los pacientes que responden a los corticosteroides (CCS); sin embargo, las recaídas son comunes y a veces es necesario administrarlos por tiempos prolongados o cambiar a otro tratamiento (96).
Si se requieren dosis de prednisona mayores a 10 mg/d de mantenimiento o las recaídas son muy constantes, hidroxiurea o interferón alfa son opciones que se puede adicionar (97) y para los pacientes que no responden a CSS o deben ser suspendidos por sus efectos adversos ciertas moléculas, como ciclosporina A, mepolizumab, alemtuzumab, reslizumab y recientemente el anticuerpo contra la cadena alfa del receptor de la IL-5 (MMEDI-563), han demostrado ser beneficiosas (50, 98-104).
El imatinib, un inhibidor selectivo de receptores tirosina quinasa, constituye la primera línea de manejo en quienes se identifica la mutación F/P y en los pacientes con características mieloproliferativas (105, 106), en quienes se han reportado la remisión de la positividad en la mutación F/P, reducción de la triptasa sérica, desaparición de las células atípicas circulantes y la mielofibrosis.
Es indispensable el uso temprano del medicamento para evitar la fibrosis tisular, pero debe tenerse precaución con la ocurrencia de miocarditis aguda eosinofílica, por lo que se deben medir troponinas antes de iniciar y, si están elevadas, usar concomitantemente con CCS, con disminución gradual según respuesta.
Se han descrito casos de pacientes con resistencia a imatinib, en los que se han identificado otras mutaciones en el gen que codifica para el PDGFRA, en donde se vienen estudiando como alternativa terapéutica, otros inhibidores de la tirosina quinasa, como desatinib y nilotinib (107).
La fotoquimioterapia extracorpórea (fototerapia), que suprime los clones deLT responsables de entidades como el linfoma cutáneo de células T, dermatitis atópica y la enfermedad de injerto versus huésped, está surgiendo como propuesta para la variante linfocítica (97, 108).
El trasplante alogénico no mieloablativo de precursores hematopoyéticos sería la última opción en pacientes que no han respondido, a pesar de todos los tratamientos disponibles(109, 110).
Conclusiones
El síndrome hipereosinofílico comprende un grupoheterogéneo de enfermedades con presentaciones clínicas muy variadas; para algunos autores el síndrome de Churg-Strauss o más recientemente conocido como granulomatosiseosinofílica con poliangiítis (83, 89, 91,111), hace parte de este gran síndrome, pero para otros deben considerarse comoentidades aparte para evitar confusiones. Hasta el momento no existen consensos globales, pero sin duda son entidades que tienen mucho en común en donde es indispensable tenerlos en mente a la hora de hacer un diagnóstico diferencial.
Agradecimientos
Agradecemos el apoyo dado de la hematóloga pediatra la doctora Natalia Builes.
Conflictos de interés
Declaramos no haber recibido ningún tipo de financiación para esta revisión y no tener conflicto de intereses.
Referencias
1. Roufosse F, Cogan E, Goldman M. Recent advances in pathogenesis and management of hypereosinophilic syndromes. Allergy 2004; 59 (7): 673-89. [ Links ]
2. Plötz SG, Hüttig B, Aigner B, Merkel C, Brockow K, Akdis C, et al. Clinical overview of cutaneous features in hypereosinophilic syndrome. Curr Allergy Asthma Rep 2012; 12 (2): 85-98. [ Links ]
3. Hardy WR, Anderson RE. The hypereosinophilic syndromes. Ann Intern Med 1968; 68 (6): 1220-9. [ Links ]
4. Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine 1975; 54 (1):1-27. [ Links ]
5. Klion AD, Bochner BS, Gleich GJ, Nutman TB, Rothenberg ME, Simon HU,et al. Approaches to the treatment of hypereosinophilic syndromes: a workshop summary report. J Allergy Clin Immunol. 2006; 117 (6): 1292-302. [ Links ]
6. Peen E, Hahn P, Lauwers G, Williams RC, Gleich G, Kephart GM. Churg-Strauss syndrome: localization of eosinophil major basic protein in damaged tissues. Arthritis Rheum 2000; 43 (8): 1897-900. [ Links ]
7. Yamada Y, Cancelas JA. FIP1L1/PDGFR alpha-associated systemic mastocytosis. Int Arch Allergy Immunol 2010; 152 (Suppl 1): 101-5. [ Links ]
8. Gleich GJ, Leiferman KM. The hypereosinophilic syndromes: current concepts and treatments. Br J Haematol 2009; 145 (3): 271-85. [ Links ]
9. Banerji A, Weller PF, Sheikh J. Cytokine-associated angioedema syndromes including episodic angioedema with eosinophilia (Gleich's Syndrome). Immunol Allergy Clin North Am 2006; 26 (4): 769-81. [ Links ]
10. Jawairia M, Shahzad G, Singh J, Rizvon K, Mustacchia P. A case report of eosinophilic esophagitis accompanying hypereosinophilic syndrome. Case RepGastrointest Med 2012; 2012: 683-572. [ Links ]
11. Lee J, Dierkhising R, Wu TT, Alexander J, Weiler C. Eosinophilic gastroin testinal disorders (EGID) with peripheral eosinophilia: a retrospective review at Mayo Clinic. Dig Dis Sci 2011; 56 (11): 3254-61. [ Links ]
12. Akuthota P, Weller PF. Eosinophilic pneumonias. Clin Microbiol Rev 2012;25 (4): 649-60. [ Links ]
13. Tsuji Y, Kawashima T, Yokota K, Tateishi Y, Kobayashi H, Itoh A, et al. Wells' syndrome as a manifestation of hypereosinophilic syndrome. Br J Dermatol 2002; 147 (4): 811-2. [ Links ]
14. Powell J, Kaur M, Muc R, Colloby P, Salim A. Persistent hypereosinophiliawith Wells syndrome. Clin Exp Dermatol 2013; 38 (1): 40-3. [ Links ]
15. Weller PF, Bubley GJ. The idiopathic hypereosinophilic syndrome. Blood 1994; 83 (10): 2759-79. [ Links ]
16. Simon D, Simon HU. Eosinophilic disorders. J Allergy Clin Immunol 2007; 119 (6): 1291-300; quiz 301-2. [ Links ]
17. Simon HU, Rothenberg ME, Bochner BS, Weller PF, Wardlaw AJ, Wechsler ME, et al. Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol 2010; 126 (1): 45-9. [ Links ]
18. Gotlib J. World Health Organization-defined eosinophilic disorders: 2012 updateon diagnosis, risk stratification, and management. Am J Hematol 2012; 87 (9): 903-14. [ Links ]
19. Rothenberg ME. Eosinophilia. N Engl J Med 1998; 338 (22): 1592-600. [ Links ]
20. Tefferi A, Patnaik MM, Pardanani A. Eosinophilia: secondary, clonal and idiopathic. Br J Haematol 2006; 133 (5): 468-92. [ Links ]
21. Brigden ML. A practical workup for eosinophilia. You can investigate the most likely causes right in your office. Postgrad Med 1999; 105 (3): 193-4, 9-202,7-10. [ Links ]
22. Campuzano G. Eosinofilia: las 215 causas mas frecuentes. Medicina y Laboratorio 2005; 11 (7/8): 321. [ Links ]
23. Brito-Babapulle F. The eosinophilias, including the idiopathic hypereosinophilic syndrome. Br J Haematol 2003; 121 (2): 203-23. [ Links ]
24. Liacouras CA, Furuta GT, Hirano I, Atkins D, Attwood SE, Bonis PA, et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol 2011; 128 (1): 3-20.e6; quiz 1-2. [ Links ]
25. Valent P, Horny HP, Bochner BS, Haferlach T, Reiter A. Controversies and open questions in the definitions and classification of the hypereosinophilic syndromes and eosinophilic leukemias. Semin Hematol 2012; 49 (2): 171-81. [ Links ]
26. Crane MM, Chang CM, Kobayashi MG, Weller PF. Incidence of myeloproliferative hypereosinophilic syndrome in the United States and an estimate of all hypereosinophilic syndrome incidence. J Allergy Clin Immunol 2010; 126 (1): 179-81. [ Links ]
27. Rothenberg ME, Klion AD, Roufosse FE, Kahn JE, Weller PF, Simon HU, et al. Treatment of patients with the hypereosinophilic syndrome with mepolizumab. N Engl J Med 2008; 358 (12): 1215-28. [ Links ]
28. Ogbogu PU, Bochner BS, Butterfield JH, Gleich GJ, Huss-Marp J, Kahn JE, et al. Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol 2009;124 (6): 1319-25.e3. [ Links ]
29. Klion AD, Noel P, Akin C, Law MA, Gilliland DG, Cools J, et al. Elevated serum tryptase levels identify a subset of patients with a myeloproliferative variant of idiopathic hypereosinophilic syndrome associated with tissue fibrosis, poor prognosis, and imatinib responsiveness. Blood 2003; 101 (12): 4660-6. [ Links ]
30. Slungaard A, Vercellotti GM, Tran T, Gleich GJ, Key NS. Eosinophil cationicgranule proteins impair thrombomodulin function. A potential mechanism for thromboembolism in hypereosinophilic heart disease. J Clin Invest 1993; 91 (4): 1721-30. [ Links ]
31. Neves JS, Weller PF. Functional extracellular eosinophil granules: novel implications in eosinophil immunobiology. Curr Opin Immunol 2009; 21 (6): 694-9. [ Links ]
32. Melo RC, Spencer LA, Dvorak AM, Weller PF. Mechanisms of eosinophilsecretion: large vesiculotubular carriers mediate transport and release of granulederived cytokines and other proteins. J Leukoc Biol 2008; 83 (2): 229-36. [ Links ]
33. Bochner BS, Gleich GJ. What targeting eosinophils has taught us about their role in diseases. J Allergy Clin Immunol 2010; 126 (1): 16-25; quiz 6-7. [ Links ]
34. Melo RC, Spencer LA, Perez SA, Neves JS, Bafford SP, Morgan ES, et al. Vesicle-mediated secretion of human eosinophil granule-derived major basic protein. Lab Invest 2009; 89 (7): 769-81. [ Links ]
35. Simon HU, Plötz S, Simon D, Seitzer U, Braathen LR, Menz G, et al. Interleukin-2 primes eosinophil degranulation in hypereosinophilia and Wells' syndrome. Eur J Immunol 2003; 33 (4): 834-9. [ Links ]
36. Wright BL, Leiferman KM, Gleich GJ. Eosinophil granule protein localization in eosinophilic endomyocardial disease. N Engl J Med 2011; 365 (2): 187-8. [ Links ]
37. Plager DA, Davis MD, Andrews AG, Coenen MJ, George TJ, Gleich GJ, et al. Eosinophil ribonucleases and their cutaneous lesion-forming activity. J Immunol 2009; 183 (6): 4013-20. [ Links ]
38. Fauci AS, Harley JB, Roberts WC, Ferrans VJ, Gralnick HR, Bjornson BH. NIH conference. The idiopathic hypereosinophilic syndrome. Clinical, pathophysiologic, and therapeutic considerations. Ann Intern Med 1982; 97 (1): 78-92. [ Links ]
39. Cogan E, Roufosse F. Clinical management of the hypereosinophilic syndromes. Expert Rev Hematol 2012; 5 (3): 275-89; quiz 90. [ Links ]
40. Helbig G, Wiśniewska-Piąty K, Francuz T, Dziaczkowska-Suszek J, Kyrcz Krzemień S. Diversity of clinical manifestations and response to corticosteroids for idiopathic hypereosinophilic syndrome: retrospective study in 33 patients. Leuk Lymphoma. 2012. [ Links ]
41. Roufosse F, Weller PF. Practical approach to the patient with hypereosinophilia. J Allergy Clin Immunol 2010; 126 (1): 39-44. [ Links ]
42. D'Orazio J, Pulliam J. Hypereosinophilic syndrome presenting as acute myocardial infarction in an adolescent. J Pediatr 2011; 158 (4): 685. [ Links ]
43. Klion A. Hypereosinophilic syndrome: current approach to diagnosis and treatment. Annu Rev Med 2009; 60: 293-306. [ Links ]
44. Minola E, Sonzogni A. Chronic hepatitis in hypereosinophilic syndrome: reportof an unusual case. Infez Med 2005; 13 (3): 182-6. [ Links ]
45. Jeon YW, Hong SJ, Kim HJ, Han JP, Kim HK, Ko BM, et al. A hypereosinophilic syndrome presenting as eosinophilic colitis. Clin Endosc 2012; 45 (4):444-7. [ Links ]
46. Blanchard C, Rothenberg ME. Biology of the eosinophil. Adv Immunol 2009; 101: 81-121. [ Links ]
47. Pardal-Fernández JM, Sáez-Méndez L, Rodríguez-Vázquez M, Godes-Medrano B, Iñíguez-De Onzoño L. [Multiple mononeuritis and eosinophilic fasciitis in a patient with idiopathic hypereosinophilic syndrome]. Rev Neurol 2012; 54 (2): 100-4. [ Links ]
48. Titlić M, Kodzoman K, Loncar D. Neurologic manifestations of hypereosinophilic syndrome--review of the literature. Acta Clin Croat 2012; 51 (1): 65-9. [ Links ]
49. Sheikh J, Weller PF. Clinical overview of hypereosinophilic syndromes. Immunol Allergy Clin North Am 2007; 27 (3): 333-55. [ Links ]
50. Samitas K, Rådinger M, Bossios A. Current update on eosinophilic lung diseases and anti-IL-5 treatment. Recent Pat Antiinfect Drug Discov 2011; 6 (3): 189-205. [ Links ]
51. Campos LE, Pereira LF. Pulmonary eosinophilia. J Bras Pneumol 2009; 35 (6): 561-73. [ Links ]
52. Barker B, Moudgil H, Slocombe G, Srinivasan K. Persistent cough: an unusualcause. Idiopathic hypereosinophilic syndrome (HES). Thorax 2010; 65 (11): 1009, 24. [ Links ]
53. Cools J, De Angelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003; 348 (13): 1201-14. [ Links ]
54. Gotlib J, Cools J, Malone JM, Schrier SL, Gilliland DG, Coutré SE. The FIP1L1-PDGFRalpha fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia: implications for diagnosis, classification, and management. Blood 2004; 103 (8): 2879-91. [ Links ]
55. Rapanotti MC, Caruso R, Ammatuna E, Zaza S, Trotta L, Divona M, et al. Molecular characterization of paediatric idiopathic hypereosinophilia. Br J Haematol 2010; 151 (5): 440-6. [ Links ]
56. Brown NJ, Stein RS. Idiopathic hypereosinophilic syndrome progressing to acute myelomonocytic leukemia with chloromas. South Med J 1989; 82 (10):1303-5. [ Links ]
57. Brito-Babapulle F. Clonal eosinophilic disorders and the hypereosinophilic syndrome. Blood Rev 1997; 11 (3): 129-45. [ Links ]
58. Ionescu MA, Wang L, Janin A. Hypereosinophilic syndrome and proliferative diseases. Acta Dermatovenerol Croat 2009; 17 (4): 323-30. [ Links ]
59. Tefferi A, Gotlib J, Pardanani A. Hypereosinophilic syndrome and clonal eosinophilia: point-of-care diagnostic algorithm and treatment update. MayoClin Proc 2010; 85 (2): 158-64. [ Links ]
60. Isvy-Joubert A, Hickman G, Flageul B, Battistella M, Bagot M, Galicier L, et al. [Myeloproliferative hypereosinophilic syndrome revealed by bipolar mucosal ulcerations]. Ann Dermatol Venereol 2012; 139 (10): 636-40. [ Links ]
61. Cottin V, Cordier JF. Eosinophilic lung diseases. Immunol Allergy Clin North Am 2012; 32 (4): 557-86. [ Links ]
62. Wang L, Wei L, Wang JC, Liu YH, Deng YC. Idiopathic hypereosinophilic syndrome revealed by encephalopathy. J Clin Neurosci 2012; 19 (12): 1746-8. [ Links ]
63. Kahn JE, Blétry O, Guillevin L. Hypereosinophilic syndromes. Best Pract Res Clin Rheumatol 2008; 22 (5): 863-82. [ Links ]
64. Brugnoni D, Airó P, Rossi G, Bettinardi A, Simon HU, Garza L, et al. A case of hypereosinophilic syndrome is associated with the expansion of a CD3-CD4+T-cell population able to secrete large amounts of interleukin-5. Blood 1996; 87 (4): 1416-22. [ Links ]
65. Simon HU, Yousefi S, Dommann-Scherrer CC, Zimmermann DR, Bauer S, Barandun J, et al. Expansion of cytokine-producing CD4-CD8- T cells associated with abnormal Fas expression and hypereosinophilia. J Exp Med 1996; 183 (3): 1071-82. [ Links ]
66. de Lavareille A, Roufosse F, Schmid-Grendelmeier P, Roumier AS, Schandené L, Cogan E, et al. High serum thymus and activation-regulated chemokinelevels in the lymphocytic variant of the hypereosinophilic syndrome. J Allergy Clin Immunol 2002; 110 (3): 476-9. [ Links ]
67. Roufosse F. Hypereosinophilic syndrome variants: diagnostic and therapeutic considerations. Haematologica 2009; 94 (9): 1188-93. [ Links ]
68. Helbig G, Wieczorkiewicz A, Dziaczkowska-Suszek J, Majewski M, Kyrcz-Krzemien S. T-cell abnormalities are present at high frequencies in patients withhypereosinophilic syndrome. Haematologica 2009; 94 (9): 1236-41. [ Links ]
69. Cogan E, Schandené L, Crusiaux A, Cochaux P, Velu T, Goldman M. Brief report: clonal proliferation of type 2 helper T cells in a man with the hypereosinophilic syndrome. N Engl J Med 1994; 330 (8): 535-8. [ Links ]
70. Roufosse F, Cogan E, Goldman M. Lymphocytic variant hypereosinophilic syndromes. Immunol Allergy Clin North Am 2007; 27 (3): 389-413. [ Links ]
71. Levesque H, Elie-Legrand MC, Thorel JM, Touchais O, Gancel A, Hecketsweiller P, et al. [Idiopathic hypereosinophilic syndrome with predominant digestive manifestations or eosinophilic gastroenteritis? Apropos of 2 cases]. Gastroenterol Clin Biol 1990; 14 (6-7): 586-8. [ Links ]
72. Scheurlen M, Mörk H, Weber P. Hypereosinophilic syndrome resembling chronic inflammatory bowel disease with primary sclerosing cholangitis. J Clin Gastroenterol 1992; 14 (1): 59-63. [ Links ]
73. Takekawa M, Imai K, Adachi M, Aoki S, Maeda K, Hinoda Y, et al. Hypereosinophilic syndrome accompanied with necrosis of finger tips. Intern Med 1992; 31 (11): 1262-6. [ Links ]
74. Trüeb RM, Lübbe J, Torricelli R, Panizzon RG, Wüthrich B, Burg G. Eosinophilic myositis with eosinophilic cellulitislike skin lesions. Association with increased serum levels of eosinophil cationic protein and interleukin-5. Arch Dermatol 1997; 133 (2): 203-6. [ Links ]
75. Trüeb RM, Pericin M, Winzeler B, Wüthrich B, Burg G. Eosinophilic myositis/perimyositis: frequency and spectrum of cutaneous manifestations. J Am Acad Dermatol 1997; 37 (3 Pt 1): 385-91. [ Links ]
76. Monaco S, Lucci B, Laperchia N, Tezzon F, Currò-Dossi B, Nardelli E, et al. Polyneuropathy in hypereosinophilic syndrome. Neurology 1988; 38 (3):494-6. [ Links ]
77. Moore PM, Harley JB, Fauci AS. Neurologic dysfunction in the idiopathic hypereosinophilic syndrome. Ann Intern Med 1985; 102 (1): 109-14. [ Links ]
78. Rapanotti MC, Caruso R, Bernardini S, Coletti V, Lo-Coco F, De Rossi G. Idiopathic hypereosinophilic syndrome: a case evolving in B-lymphoblastic lymphoma. Leuk Res 2005; 29 (8): 975-9. [ Links ]
79. Roufosse F, Schandené L, Sibille C, Willard-Gallo K, Kennes B, Efira A, et al. Clonal Th2 lymphocytes in patients with the idiopathic hypereosinophilic syndrome. Br J Haematol 2000; 109 (3): 540-8. [ Links ]
80. Bernasconi C. Hypereosinophilia in acute lymphoblastic leukemia: an association to be remembered. Haematologica 2001; 86 (8): 893. [ Links ]
81. Della-Torre E, Tresoldi M, Scotti R, Praderio L, Mellone R, Ponzoni M, et al. Atypical presentation of Churg-Strauss syndrome or an undescribed hypereosinophilic disease? J Allergy Clin Immunol 2011; 128 (4): 908-11. [ Links ]
82. Terrier B, Bièche I, Maisonobe T, Laurendeau I, Rosenzwajg M, Kahn JE, et al. Interleukin-25: a cytokine linking eosinophils and adaptive immunity in Churg-Strauss syndrome. Blood 2010; 116 (22): 4523-31. [ Links ]
83. Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis Rheum. 2013; 65 (1): 270-81. [ Links ]
84. Holle JU, Moosig F, Gross WL. Diagnostic and therapeutic management of Churg-Strauss syndrome. Expert Rev Clin Immunol 2009; 5 (6): 813-23. [ Links ]
85. Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) 1999; 78 (1): 26-37. [ Links ]
86. Baldini C, Talarico R, Della Rossa A, Bombardieri S. Clinical manifestations and treatment of Churg-Strauss syndrome. Rheum Dis Clin North Am 2010; 36 (3): 527-43. [ Links ]
87. Khoury P, Zagallo P, Talar-Williams C, Santos CS, Dinerman E, Holland NC, et al. Serum biomarkers are similar in Churg-Strauss syndrome and hypereosinophilic syndrome. Allergy 2012; 67 (9): 1149-56. [ Links ]
88. Pagnoux C, Guillevin L. Churg-Strauss syndrome: evidence for disease subtypes? Curr Opin Rheumatol 2010; 22 (1): 21-8. [ Links ]
89. Abril A, Calamia KT, Cohen MD. The Churg Strauss syndrome (allergic granulomatous angiitis): review and update. Semin Arthritis Rheum 2003; 33 (2): 106-14. [ Links ]
90. Pugliese N, Bruzzone M, Della Rossa A, Baldini C, Catarsi E, Tavoni A, et al. [Churg-Strauss vasculitis and idiopathic hypereosinophyl syndrome: role of molecular biology in the differential diagnosis of hypereosinophyl syndrome]. Reumatismo 2008; 60 (2): 120-4. [ Links ]
91. Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990;33 (8): 1094-100. [ Links ]
92. Abril A. Churg-strauss syndrome: an update. Curr Rheumatol Rep 2011; 13 (6): 489-95. [ Links ]
93. Valent P, Klion AD, Horny HP, Roufosse F, Gotlib J, Weller PF, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol 2012; 130 (3): 607-12. e9. [ Links ]
94. Butterfield JH, Weiler CR. Treatment of hypereosinophilic syndromes--the first 100 years. Semin Hematol 2012; 49 (2): 182-91. [ Links ]
95. Sheikh J, Weller PF. Advances in diagnosis and treatment of eosinophilia. Curr Opin Hematol 2009; 16 (1): 3-8. [ Links ]
96. Christoforidou A, Kotsianidis I, Margaritis D, Anastasiadis A, Douvali E, Tsatalas C. Long-term remission of lymphocytic hypereosinophilic syndrome with imatinib mesylate. Am J Hematol 2012; 87 (1): 131-2. [ Links ]
97. Wechsler ME, Fulkerson PC, Bochner BS, Gauvreau GM, Gleich GJ, Henkel T, et al. Novel targeted therapies for eosinophilic disorders. J Allergy Clin Immunol 2012; 130 (3): 563-71. [ Links ]
98. Corren J. Inhibition of interleukin-5 for the treatment of eosinophilic diseases. Discov Med 2012; 13 (71): 305-12. [ Links ]
99. Abonia JP, Putnam PE. Mepolizumab in eosinophilic disorders. Expert Rev Clin Immunol 2011; 7 (4): 411-7. [ Links ]
100. Kim S, Marigowda G, Oren E, Israel E, Wechsler ME. Mepolizumab as a steroid-sparing treatment option in patients with Churg-Strauss syndrome. J Allergy Clin Immunol 2010; 125 (6): 1336-43. [ Links ]
101. Srinivasan A, Lavanya R, Sankar J. Steroid-unresponsive hypereosinophilic syndrome. Ann Trop Paediatr 2011; 31 (3): 273-7. [ Links ]
102. Schwartz LB, Sheikh J, Singh A. Current strategies in the management of hypereosinophilic syndrome, including mepolizumab. Curr Med Res Opin 2010;26 (8): 1933-46. [ Links ]
103. Wagner LA, Speckart S, Cutter B, Gleich GJ. Treatment of FIP1L1/PDGFRA negative hypereosinophilic syndrome with alemtuzumab, an anti-CD52 antibody. J Allergy Clin Immunol 2009; 123 (6): 1407-8. [ Links ]
104. Strati P, Cortes J, Faderl S, Kantarjian H, Verstovsek S. Long-term followup of patients with hypereosinophilic syndrome treated with Alemtuzumab, an anti-CD52 antibody. Clin Lymphoma Myeloma Leuk 2013; 13 (3): 287-91. [ Links ]
105. Arefi M, García JL, Briz MM, de Arriba F, Rodríguez JN, Martín-Núñez G,et al. Response to imatinib mesylate in patients with hypereosinophilic syndrome. Int J Hematol 2012; 96 (3): 320-6. [ Links ]
106. Helbig G, Hus M, Hałasz M, Dudziński M, Więcławek A, Stachowicz M, et al. Imatinib mesylate may induce long-term clinical response in FIP1L1-PDGFRα negative hypereosinophilic syndrome. Med Oncol 2012; 29 (2): 1073-6. [ Links ]
107. Ikezoe T, Togitani K, Tasaka T, Nishioka C, Yokoyama A. Successful treatment of imatinib-resistant hypereosinophilic syndrome with nilotinib. Leuk Res 2010; 34 (8): e200-1. [ Links ]
108. Gattringer C, Müller H, Steurer M, Steger C, Ratzinger G. Narrowband UVB therapy for the treatment of pruritus in hypereosinophilic syndrome: clinical report and review of the literature on phototherapy. J Am Acad Dermatol 2012;67 (5): e210-3. [ Links ]
109. Baccarani M, Deininger MW, Rosti G, Hochhaus A, Soverini S, Apperley JF, et al. European Leukemia Net recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013; 122 (6): 872-84. [ Links ]
110. Roufosse F, Cogan E, Goldman M. The hypereosinophilic syndrome revisited. Annu Rev Med 2003; 54: 169-84. [ Links ]
111. Vaglio A, Buzio C, Zwerina J. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): state of the art. Allergy 2013; 68 (3): 261-73. [ Links ]
