










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActa Medica Colombiana
versión impresa ISSN 0120-2448
Acta Med Colomb vol.39 no.3 Bogotá jul./sep. 2014
Presentación de casos
Polimiositis y compromiso cardiaco
Cardiac involvement in polymyositis
July Andrea Russi1, Agustín Paz1, Jaime Valdés1, Douglas Rodríguez1, Julián Valencia1, Guillermo mora2
1 Residentes de Medicina Interna, Departamento Medicina Interna, Facultad de Medicina, Universidad Nacional de Colombia. Bogotá, D.C. (Colombia).
2 Cardiólogo-Electrofisiólogo, Docente, Departamento Medicina Interna, Facultad de Medicina, Universidad Nacional de Colombia. Bogotá, D.C. (Colombia).
Correspondencia. Dr. Guillermo Mora Pabón. E-mail: gmorap@unal.edu.co
Recibido: 22/VII/2013Aceptado: 25/VIII/2014
Resumen
Las miopatías idiopáticas son enfermedades que se caracterizan por cursar con una lesión muscula de tipo inflamatorio, sin embargo, también se ha encontrado que presentan compromiso del músculo cardiaco de forma importante, inclusive con desarrollo de falla cardiaca. Lo anterior constituye una causa frecuente de morbilidad y mortalidad en este grupo de pacientes. Al respecto, se presenta el caso de una paciente de 54 años con sospecha inicial de síndrome coronario agudo, quien cursó con polimiositis, dolor torácico y compromiso de la fracción de eyección.
Palabras clave: polimiositis, cardiomiopatía.
Abstract
Idiopathic myopathies are disorders characterized by inflammatory muscle damage. However, it has also been found that they may present significant muscle heart engagement, even with the development of heart failure. This is a frequent cause of morbidity and mortality in this group of patients. In this regard, the case of a 54 year old female patient with initial suspicion of acute coronary syndrome accompanied by polymyositis, chest pain and commitment of the ejection fraction, is presented.
Key words: cardiomyopathies, polymyositis.
Introducción
La polimiositis hace parte del heterogéneo grupo de las miopatías inflamatorias (polimiositis, dermatomiositis y miositis por cuerpos de inclusión), la mayoría de ellas de origen autoinmune que comprometen principalmente el músculo esquelético y en grados variables otros órganos y sistemas como piel, sistema cardiovascular, respiratorio y gastrointestinal. Presenta grados variables de inflamación muscular, con compromiso principalmente de la musculatura proximal (1). En comparación con la dermatomiositis, cuya forma de presentación suele ser más evidente dado el compromiso cutáneo (rash), la polimiositis suele representar un mayor reto diagnóstico, fácilmente se confunde con otros tipos de miopatía y sigue considerándose un diagnóstico de exclusión luego de descartar entre otras, afección por enfermedades neuromusculares, medicamentos miotóxicos, o endocrinopatías (2).
Aunque el compromiso cardiaco no se incluye en los criterios diagnósticos, se ha demostrado que se asocia con peor pronóstico y constituye la causa más frecuente de mortalidad (20%), relacionada con falla cardiaca, enfermedad coronaria, miocarditis y arritmias. Recientemente gracias a la aparición de métodos diagnósticos más sensibles y menos invasivos se ha logrado la identificación de diferentes formas de compromiso cardiaco desde presentaciones subclínicas hasta casos fatales. En un estudio de cohorte con seguimiento a seis años se encontró que pacientes con polimiositis tenían aumento de la mortalidad hasta de cuatro veces, principalmente relacionada con el desarrollo de infarto agudo de miocardio (3).
Presentación del caso
Mujer de 54 años procedente de Fusagasugá (Cundinamarca) que ingresa al servicio de urgencias remitida de la consulta externa de reumatología por sospecha de miopatía con cuadro de tres años de evolución de astenia, adinamia dolor muscular principalmente en la cintura escapular y cadera con exacerbaciones y remisiones, negaba regurgitación, disfagia o síntomas mucocutáneos. Tres meses antes, presentó dolor torácico opresivo constante no asociado a esfuerzo, acompañado de palpitaciones, motivo por el cual recibió atención médica en otra institución, donde con la impresión de síndrome coronario agudo realizaron cateterismo cardiaco encontrando coronarias epicárdicas sanas.
Tiene antecedentes de cáncer de cérvix hace 20 años tratado con histerectomía en remisión, hipertensión arterial controlada, gastritis crónica activa (biopsia reporta metaplasia intestinal tipo colónico sin malignidad). Niega antecedentes tóxicos. Recibía prednisolona 20 mg día, enalapril 5 mg día, metoprolol 50 mg/12 horas. Al examen físico tenía ruidos cardiacos sin soplos, ni extratonos, disminución generalizada de la fuerza muscular simétrica de predominio proximal de 3/5 en las cuatro extremidades, reflejos osteotendinosos simétricos, sin déficit neurológico focal.
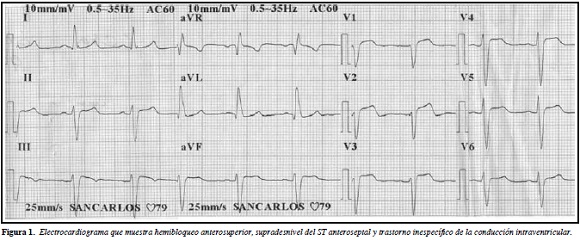
Los paraclínicos iniciales (en el momento del dolor torácico) muestran electrocardiograma con hemibloqueo anterosuperior, supradesnivel del segmento ST en cara anterior y QRS 110 ms. Creatinkinasa (CK) total 1152 U/L con posterior descenso progresivo hasta 366 U/L, CK-MB 183 U/L, troponina negativa seriada. Ecocardiograma con hipertrofia ventrículo izquierdo leve, fracción de eyección del ventrículo izquierdo (FEVI) 55%, trastorno segmentario contractilidad leve del segmento apical de la pared anterior y septum, disfunción diastólica y leve dilatación biauricular. Anticuerpos antinucleares dilución 1/640 patrón centrómero (Método IFI-Células Hep II), antiRo SSA 2.85 UE/mL, antiSm 3.38 E/mL, antiRNP 4.2UE-mL, anti Scl70 3.4, Aldolasa 23.3UI/L todos los anteriores por método inmunoenzimático - EIA.
Durante la hospitalización, se inicia estudio para descartar etiología paraneoplásica con ecografía abdominal, radiografía y tomografía de tórax, colonoscopia, antígeno carcinoembrionario y alfafeto proteína los cuales mostraron resultados dentro de los rangos de referencia. No presentaba alteración en ninguna de las líneas celulares del cuadro hemático, ni en la función renal. CK total de ingreso en 427 U/L y elevación de transaminasas (AST 36 UI/L, ALT 44 UI/L), sin otra alteración en perfil hepático. Las pruebas de neuroconducción fueron normales, la electromiografía mostró signos de denervación dados por potenciales agudos positivos y micropotenciales de fibrilación con ondas motoras de franco patrón miopático, disminuidas en frecuencia, sugestivos de enfermedad inflamatoria leve de la fibramuscular (polimiositis).
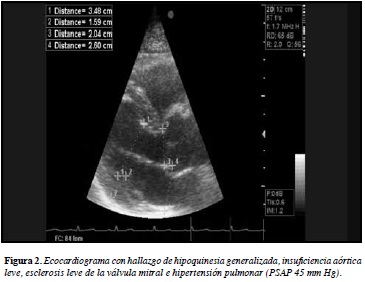
La paciente persistió con dolor torácico y con electrocardiogramas sin cambios dinámicos (Figura 1) y ecocardiograma de control con FEVI comprometida 40% (Figura 2).
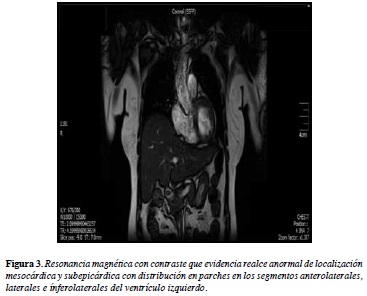
Se aumentó la dosis de prednisolona a 50 mg/día (que ya recibía previamente) y se inició metotrexate 15 mg semanales, con desparasitación y suplemento con ácido fólico. Luego de tres días de la nueva hospitalización, la paciente inicia lenta mejoría de la fuerza muscular y disminución de la CK total a 272 U/L. Posteriormente en el seguimiento ambulatorio a los dos meses de tratamiento en abril 2013, se realizó una resonancia magnética cardiaca con contraste, encontrando una FEVI de 50% con hipoquinesia de la pared lateral del ventrículo izquierdo con realce, sin derrame, hallazgos compatibles con infiltración miocárdica de origen autoinmune (Figura 3). La fuerza muscular era normal. La biopsia muscular de ese mismo mes se encontró fascículos musculares de histología normal; sin embargo, ésta se realizó después de iniciado el tratamiento.
Discusión
La polimiositis es una entidad poco frecuente, su incidencia aumenta luego de los 20 años de edad a aproximadamente 4-10 casos por millón/año y afecta principalmente a las mujeres en una proporción de 2-3:1 similar a la mayoría de enfermedades autoinmunes (4). Actualmente, siguen utilizándose los criterios diagnósticos de Bohan y Peter que utilizan variables clínicas (debilidad muscular simétrica proximal), de laboratorio (elevación de enzimas musculares), electromiográficas (potenciales motores polifásicos, cortos y pequeños, fibrilación, ondas agudas positivas, aumento de la irritabilidad de inserción y descargas repetitivas de alta frecuencia), histológicas (degeneración, regeneración, necrosis e infiltrados intersticiales mononucleares) y características mucocutáneas de dermatomiositis (eritema heliotropo periocular y signo de Gottron) (5). Los criterios no incluyen el compromiso miocárdico. De acuerdo con estos criterios se considera que el diagnóstico es definitivo si cumple con cuatro de los cinco, probable si cumple con tres y posible si cumple con dos criterios. La sensibilidad global de estos criterios varían entre 74-100%, mientras que su especificidad es 90%, cuando se compara con otras enfermedades autoinmunes sistémicas (6).
El compromiso cardiaco fue descrito por primera vez en 1899 (3), pero sólo en las últimas décadas, se ha podido documentar dicho compromiso de forma más consistente, gracias a la aparición de métodos diagnósticos más sensibles y menos invasivos, que permiten la identificación de diferentes formas, desde presentaciones subclínicas hasta casos fatales. No hay claridad de la frecuencia del compromiso cardiaco, ya que no se cuenta con criterios establecidos, pero varía entre 6 y 75%, siendo mayor en los estudios diseñados para la búsqueda del mismo. En general, es más probable el compromiso cardiaco en la polimiositis que en la dermatomiositis, encontrándose que ciertos tipos de autoanticuerpos como los antiRo se han asociado con lesión miocárdica (7).
Las manifestaciones clínicas de compromiso cardiaco son infrecuentes y son secundarias a miocarditis con un proceso inflamatorio similar al presentado en el músculoesquelético, relacionándose con mal pronóstico (7) con mortalidad de 10-20% (3). Se caracteriza por fibrosis focal, vasculitis, proliferación intimal y esclerosis de la capa media vascular e infiltración linfocítica (7). El rendimiento diagnóstico de la biopsia miocárdica depende del sitio del músculo cardiaco tomado para el estudio histopatológico, al respecto, las biopsias obtenidas de diferentes partes del ventrículo derecho pueden o no mostrar infiltrados focales (variabilidad espacial), debido a la naturaleza irregular de las lesiones en el miocardio, y las biopsias obtenidas en diferentes momentos en el tiempo documentan infiltrados inflamatorios intermitentes (variabilidad temporal) (8). Los síntomas más usuales son disnea (10%), angina (7%) y edema(3%). La falla cardiaca es la entidad clínica más observada (32-77%), seguida por las arritmias (13.8%). Esta enfermedad puede cursar también con pericarditis, hipertensión pulmonar, miocardiopatía hipertrófica sin correlacionarse con la severidad del compromiso del músculo esquelético (9).
La cardiopatía puede presentarse en cualquier estadio de la enfermedad, incluso durante su remisión. Estudios longitudinales han observado progresión de cardiopatía y aparición de nuevas anormalidades a pesar de manejo con esteroides y logro de remisión de la enfermedad. El compromiso subclínico se evidencia a través de anormalidades en las pruebas diagnósticas no invasivas como electrocardiograma de ritmo y de superficie, ecocardiografía y perfusión miocárdica. La incidencia de anormalidades electrocardiográficas en la mayoría de los estudios se observa entre 25 y 85% y las más comunes son las extrasístoles auriculares y ventriculares (18%), anormalidades de conducción del nodo sinusal (14%), hipertrofia ventricular izquierda (7%), bloqueos de rama, anormalidades inespecíficas del ST-T (5%), bloqueo auriculoventricular (4%), taquicardia supraventricular (3%)y fibrilación auricular (2%), entre otros (10). Nuestra paciente presentaba hemibloqueo izquierdo anterior y cambios del segmento ST en pared anterior.
Las anormalidades ecocardiográficas varían de 12-64% e incluyen cambios en la estructura cardiaca como dilatación auricular y ventricular izquierdas (8-12%), hipertrofia ventricular izquierda (8-15%), valvulopatía (7-23%) como prolapso mitral, estenosis o insuficiencia aórtica y mitral y derrame pericárdico (8-66%) que suele ser pequeño y sin repercusión hemodinámica. También son frecuentes desórdenes de la función cardiaca como disfunción diastólica (6-12%) o hipoquinesia global o segmentaria. La hipertensión pulmonar, la cual suele ser leve a moderada, se ha evidenciado hasta en 75% de los casos (9). En nuestro caso se evidenciaron cambios segmentarios de contractilidad y disfunción sistólica del ventrículo izquierdo, los cuales mejoraron con el tratamiento instaurado.
La resonancia magnética nuclear con contraste con gadolinio ha sido utilizada para la detección temprana de miocarditis y es útil en el seguimiento de pacientes bajo tratamiento para la identificación de corrección de trastornos de motilidad y reducción de las áreas de cicatrización generadas por el proceso inflamatorio (11). Nuestra paciente muestra en la resonancia nuclear cardiaca compromiso miocárdico compatible con daño por polimiositis. El realce del medio de contraste es un hallazgo frecuente en el escenario clínico de sospecha de miocarditis y está asociado con inflamación activa definida por histopatología (12).
En el caso expuesto se observa cómo la paciente presentó dolor torácico, disnea y alteraciones electrocardiográficas con compromiso de la fracción de eyección, además del trastorno diastólico de la relajación (no atribuibles a hipertensión, dada la rápida evolución de cuadro clínico) secundarios a miopatía inflamatoria (se descartó enfermedad coronaria con el cateterismo). Presentó además mejoría clínica y en la función ventricular posterior al manejo farmacológico.
Por otra parte, aunque en algunos casos la presencia de enfermedad coronaria es concomitante y se ha reportado prevalencia de isquemia miocárdica hasta en 26%, la elevación de troponina I representa afectación inflamatoria miocárdica de cualquier tipo y no tiene utilidad como marcador de enfermedad coronaria o inclusive de compromiso cardiaco en este contexto, ya que en este tipo de pacientes se ha encontrado positiva a pesar de la ausencia de enfermedad clínica significativa. Está por evaluarse su utilidad en predecir mortalidad o afectación cardiaca subclínica, como lo reportan Fisher et al. en un estudio observacional (13).
El tratamiento debe fundamentarse en el control del proceso inflamatorio de base, se han reportado casos de mejoría de falla cardiaca, arritmias y derrame pericárdico mediante el uso de ciclos de glucocorticoides, principalmente prednisolona. Sin embargo, existen algunos signos de progresión de la enfermedad a pesar de intervención farmacológica (corticoides e inmunosupresores como azatioprina, metotrexate y ciclofosfamida) (14). Aun así, éste es el pilar fundamental del tratamiento junto con el bloqueo neurohumoral para el manejo de la falla cardiaca (15). Hay reportes de casos de mejoría del compromiso cardiaco con el uso de medicamentos biológicos, como el rituximab (16).
Existen algunas particularidades en nuestro caso, una de ellas es la presencia de dolor importante en el cuadro clínico. A menudo se cree que son enfermedades no dolorosas; sin embargo, tanto la polimiositis como la dermatomiositis pueden asociarse a mialgias e hipersensibilidad muscular durante el curso temprano de la enfermedad, más frecuentemente en la dermatomiositis (17). Las mialgias y el dolora la palpación de las masas musculares ocurren entre 25%y 50% de los pacientes. No es tan dolorosa como en lapolimialgia reumática, fibromialgia o las miositis virales obacterianas (18, 19). Un estudio retrospectivo llevado a cabo en una unidad de enfermedades neuromusculares en Brasil sobre manifestaciones clínicas de la polimiositis evidenció que el dolor muscular (45.8%) era el síntoma más frecuente después de la debilidad muscular proximal (95.8%) llevando inclusive a la idea de utilizar dicho síntoma como criterio diagnóstico adicional (20).
Por último debemos notar que nuestra paciente tenía una biopsia muscular negativa. La biopsia muscular es una prueba importante para establecer el diagnóstico de la polimiositis y hace parte de los criterios de Bohan y Peter (5). Sin embargo, la inflamación en la polimiositises segmentaria, por lo que se puede encontrar un resultado normal pese a tener la enfermedad en 20% de pacientes en biopsia con aguja (21) y 17% en biopsia abierta (22). Se puede considerar la toma de biopsia de un músculodiferente si el paciente cumple los criterios clínicos, pero la primera muestra no fue diagnóstica. En algunos casos la resonancia magnética muscular puede ser de utilidad en identificar las zonas inflamatorias y seleccionar el área para tomar la biopsia (1).
Conclusiones
La polimiositis es una enfermedad reconocida frecuentemente por el compromiso muscular esquelético en la quese debe evaluar siempre la presencia de lesión miocárdicaconcomitante, debido a que esta afectación puede no producir síntomas y es considerada una de las principales causas de mortalidad en este grupo de pacientes.
Referencias
1. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet 2003; 62(9388): 971-82. [ Links ]
2. Briani C, Doria A, Sarzi-Puttini P, Dalakas MC. Update on idiopathic inflammatory myopathies. Autoimmunity 2006; 39: 161-70. [ Links ]
3. Lundberg IE. The heart in dermatomyositis and polymyositis. Rheumatology2006; 45(Suppl 4): 18-21. [ Links ]
4. Madan V, Chinoy H, Griffiths CE, Cooper RG. Defining cancer risk in dermatomyositis. Part I. Clinical and experimental dermatology 2009; 34: 451-5. [ Links ]
5. Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975; 292: 403-7. [ Links ]
6. Targoff IN, Miller FW, Medsger TA Jr, Oddis CV. Classification criteria forthe idiopathic inflammatory myopathies. Curr Opin Rheumatol 1997; 9: 527-535. [ Links ]
7. Lu Z, Guo-chun W, Li M, Ning Z. Cardiac involvement in adult polymyositis or dermatomyositis: a systematic review. Clin Cardiol 2012; 35: 686-91. [ Links ]
8. Hauck AJ, Kearney DL, Edwards WD. Evaluation of postmortemendomyocardial biopsy specimens from 38 patients with lymphocytic myocarditis:implications for role of sampling error. Mayo Clin Proc 1989; 64: 1235-1245. [ Links ]
9. Senechal M, Crete M, Couture C, Poirier P. Myocardial dysfunction in polymyositis. Can J Cardiol 2006; 22: 869-71. [ Links ]
10. Gupta R, Wayangankar SA, Targoff IN, Hennebry TA. Clinical cardiac involvement in idiopathic inflammatory myopathies: a systematic review. Intern J Cardiol 2011; 148: 261-70. [ Links ]
11. Allanore Y, Vignaux O, Arnaud L, Puechal X, Pavy S, Duboc D, et al. Effects of corticosteroids and immunosuppressors on idiopathic inflammatory myopathy related myocarditis evaluated by magnetic resonance imaging. Ann Rheum Dis 2006; 65: 249-52. [ Links ]
12. Mahrholdt H, Goedecke C, Wagner A, Meinhardt G,AthanasiadisA,Vogelsberg H, Fritz P, Klingel K, Kandolf R, Sechtem U. Cardiovascular magnetic resonance assessment of human myocarditis: a comparison to histology and molecular pathology. Circulation 2004; 109: 1250-1258. [ Links ]
13. Fisher C, Agrawal S, Wong WM, Fahie-Wilson M, Dasgupta B. Clinical observations on the significance of raised cardiac troponin-T in patients with myositis of varying etiologies seen in rheumatology practice. Clin Rheumat 2010; 29(10): 1107-11. [ Links ]
14. Odabasi Z, Yapundich R, Oh SJ. Polymyositis presenting with cardiac manifestations: Report of two cases and review of the literature. Clin Neurol Neurosurg 2010; 112: 160-3. [ Links ]
15. Hengstman GJ, van den Hoogen FH, van Engelen BG. Treatment of the inflammatory myopathies: update and practical recommendations. Exp OpinPharmacotherapy 2009; 10: 1183-90. [ Links ]
16. Touma Z,Arayssi T, Kibbi L, MasriAF. Successful treatment of cardiac involvement in dermatomyositis with rituximab. Joint, bone, spine: revue du rhumatisme 2008; 75: 334-7. [ Links ]
17. Khan S, Christopher-Stine L. Polymyositis, dermatomyositis and autoimmune necrotizing myopathy: clinical features. Rheum Dis Clin N Am 2011; 37: 143-158. [ Links ]
18. Dalakas M, Hohlfeld R. Polymyositis and dermatomyositis. Lancet 2003; 362: 971-982. [ Links ]
19. Miller M, Vleugels R. Clinical manifestations of dermatomyositis and polymyositis in adults. UpToDate. 2013. [ Links ]
20. Scola S, Werneck L. Diagnosis of dermatomyositis and polymyositis: a study of 102 cases. Arq Neuro-Psiquiatr 2000; 58: 789-99. [ Links ]
21. Ehrenstein MR, Snaith ML, Isenberg DA. Idiopathic myositis: a rheumatological view. Annals of the Rheumatic Diseases 1992; 51: 41-44. [ Links ]
22. DeVere R, Bradley W G. Polymyositis: its presentation, morbidity and mortality. Brain 1975; 98: 637-66. [ Links ]
