










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActa Medica Colombiana
versión impresa ISSN 0120-2448
Acta Med Colomb vol.40 no.1 Bogotá ene./mar. 2015
Presentación de casos
Telangiectasia hemorrágica hereditaria Síndrome de Osler Weber Rendú y manejo con bevacizumab
Hereditary hemorrhagic telangiectasia Osler Weber Rendu syndrome and management with bevacizumab
Juan Felipe Combariza, Viviana Patricia Olaya • Medellín (Colombia)
Dr. Juan Felipe Combariza: Hematólogo; Dra. Viviana Patricia Olaya: Internista. Hospital Pablo Tobón Uribe. Medellín (Colombia).
Correspondencia Dr. Juan Felipe Combariza.
Medellín (Colombia).
E-mail: combariza@yahoo.com
Recibido: 31/X/2013 Aceptado: 8/X/2014
Resumen
Presentamos un caso clínico de una paciente con diagnóstico de telangiectasia hemorrágica hereditaria con múltiples manifestaciones sistémicas debidas a sangrados profusos, anemia severa y malformaciones arteriovenosas pulmonares, hepáticas y falla cardiaca de alto débito, con adecuada respuesta al uso de bevacizumab. (Acta Med Colomb 2015; 40: 66-68).
Palabras clave: telangiectasia hemorrágica hereditaria, síndrome de Osler Weber Rendu, bevacizumab, malformaciones arteriovenosas.
Abstract
The case of a patient diagnosed with hereditary hemorrhagic telangiectasia with multiple systemic manifestations due to profuse bleeding, severe anemia and pulmonary arteriovenous malformations, liver and heart failure high debit adequate response to the use of bevacizumab, is presented. (Acta Med Colomb 2015; 40: 66-68).
Keywords: hereditary hemorrhagic telangiectasia, Osler Weber Rendu, bevacizumab, arterio-venous malformations.
Introducción
La telangiectasia hemorrágica hereditaria THH o síndrome de Osler- Weber Rendú es un desorden autosomico dominante caracterizado por telangiectasias en piel y mucosas y malformaciones arteriovenosas; clínicamente se manifiestan por un amplio espectro de signos y síntomas, la principal manifestación es sangrado mucocutáneo, usualmente epistaxis que ocurre en más de 90% de los pacientes y por sangrados recurrentes lleva a una anemia ferropénica de difícil manejo con baja respuesta con el hierro parenteral y requerimiento de soporte transfusional a repetición (1). De acuerdo con el sitio de localización de las telangiectasias o las malformaciones pueden producir otras manifestaciones como son sangrado gastrointestinal, disfunción hepática, hipertensión pulmonar.
El diagnóstico se realiza por unos criterios denominados criterios de Curaçao, (2,3) en los cuales deben cumplir por lo menos tres para realizar el diagnóstico.
Los criterios son: sangrado nasal espontáneo y recurrente, telangiectasias, historia familiar, lesiones viscerales.
En los pacientes con THH existe un aumento del factor de crecimiento del endotelio vascular y se ha realizado una hipótesis en la que un imbalance entre factores antiangiogénicos y proangiogénicos (4,5).
Por su mecanismo fisiopatológico se han descritos varios reportes de casos y series de casos del uso del bevacizumab, un inhibidor del factor de crecimiento del endotelio vascular en el manejo de esta enfermedad con disminución de los episodios de sangrado, mejoría de la anemia y en algunos casos mejoría de la función hepática.
En este momento presentamos un caso de una mujer con THH con anemia severa dependiente de transfusión asociada a malformaciones arteriovenosas hepáticas severas y pulmonar y falla cardiaca de alto gasto con hipertensión pulmonar, que recibió manejo exitoso con bevacizumab.
Caso clínico
Paciente femenina de 60 años con antecedente de THH manifiesta por sangrados nasales espontáneos diarios asociado a anemia ferropénica severa refractaria al uso de hierroparenteral y con requerimiento de soporte transfusional periódico cada cuatro a seis semanas.
Así mismo ha presentado malformaciones severas hepáticas y pulmonares requiriendo embolizaciones en varias oportunidades, ocasionando una falla cardiaca de alto gasto con hipertensión pulmonar severa, con presión sistólica de arteria pulmonar de 79 mmHg y documentando telangiectasias en manos en nariz y en mucosa oral y con antecedentes familiares hermanos y sobrinos con el mismo cuadro.
Manejo crónico con hierro oral - furosemida - enalapril, oxígeno domiciliario.
En el examen físico inicial se documentaron múltiples telangiectasias en manos y en lengua con edema grado II de miembros inferiores.
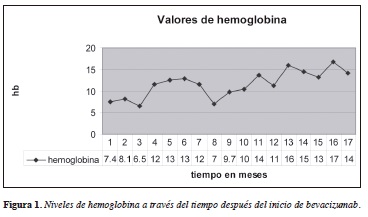
Se inició tratamiento con bevacizumab a una dosis de 5 mg/Kg cada 15 días por seis aplicaciones entre febrero y mayo de 2012 con adecuada tolerancia, con disminución importante del soporte transfusional, requiriendo sólo un episodio transfusional de glóbulos rojos. En marzo de 2012 antes de ingresar a la segunda aplicación del medicamento presentó un sangrado severo nasal con niveles de hemoglobina que disminuyeron hasta 6.5 g/dL, posterior a este episodio no hubo nuevos requerimientos transfusionales.
Evoluciona hacia la mejoría clínica con elevación progresiva de los niveles de hemoglobina; durante este periodo desde valores basales previo al inicio de 7.4 g/dL. hasta hemoglobina de 12.8 g/dL asociado a disminución de los sangrados diarios tanto en volumen como en periodicidad y mejoría de su estado funcional con disminución de la disnea, mejor tolerancia al ejercicio y disminución de los edemas.
Después de este periodo de seis aplicaciones debe vacizumab continúa en observación pero cuatro meses después de terminar el tratamiento nuevamente presenta aumento de los sangrados mucocutáneos con descenso de la hemoglobina hasta niveles de 7 g/dL, por lo cual se considera reinicio de tratamiento con bevacizumab usando la misma dosis 5 mg/Kg de manera quincenal hasta completar nuevamente seis dosis y posteriormente en diciembre de 2012 bevacizumab de mantenimiento cada tres semanas, con lo cual presenta nuevamente mejoría de los parámetros hematológicas y clínicos, disminución del sangrado, mejoría de los niveles de hemoglobina independiente de soporte transfusional y resolución completa de los edemas con mejoría a la tolerancia al ejercicio.
En junio de 2013 se disminuye la dosis de bevacizumaba 3 mg/Kg cada tres semanas, sin presentar cambios en su estado clínico ni descenso en la hemoglobina y en agosto de2013 se continúa tratamiento con la misma dosis del medicamento, pero aplicación una vez al mes, continuando con estabilidad clínica, independencia transfusional, adecuado control del sangrado y de su estado funcional, volviendo el hígado a tamaños normales, resolución completa de los edemas, adecuada tolerancia al ejercicio y tornándose independiente del oxígeno.
Al realizar evaluación imagenológica las malformaciones arteriovenosas persistían del mismo tamaño sin variaciones con respecto a los estudios basales.
En el momento la paciente completa 19 meses de seguimiento, con 18 meses libre de transfusión con nivelesde hemoglobina entre 13 y 17 g/dL durante los últimos seis meses y con adecuado control del sangrado y de las manifestaciones sistémicas dadas por las malformaciones arteriovenosas severas, durante toda su evolución nunca presentó manifestaciones de toxicidad por el medicamento reportadas en el contexto de enfermedades neoplásicas.
En la Figura 1 se evidencian los niveles de hemoglobina de acuerdo con el tiempo en meses y los requerimientos transfusionales y aplicación de bevacizumab.
Discusión
El síndrome de Osler-Weber-Rendu o telangiectasia hemorrágica hereditaria es un raro desorden de herencia autosómica dominante, que ocasiona sangrados mucocutáneos principalmente nasales diariamente, lo cual lleva a anemia ferropénica persistente, con baja respuesta a los suplementos de hierro oral y parenteral por pérdidas superiores a los administrados y requerimientos frecuentes de soporte transfusional (1).
El manejo tradicional de esta enfermedad ha sido de soporte para control de las manifestaciones de la anemia y tratamientos para disminuir la cantidad de sangrados nasales como son electrocauterio, terapia con láser, antifibrinolíticos, son poco exitosos con rápidas recaídas del sangrado, tratamiento. Cuando existe un componente de malformaciones viscerales pueden producir complicaciones mayores, como sangrados gastrointestinales, depende del sitio de localización de malformación adicionales se han descrito malformaciones hepáticas, cerebrales, pulmonares etc., también con respuestas variables al manejo de intervención con embolizaciones locales o sistémicas (6-10).
Pueden existir malformaciones hepáticas hasta en el 84%de los casos que pueden ocasionar principalmente falla cardiaca de alto gasto, hipertensión portal y en algunos casos necrosis biliar. Esta falla cardiaca de alto gasto es debido a que los shunts hepáticos así como otros shunts extracardiacos sistémicos elevan la precarga, con disminución de la resistencia vascular periférica llevando a un aumento progresivo del gasto cardiaco que aumenta la presión del ventrículo izquierdo, disnea, falla cardiaca e hipertensión pulmonar (11).
Se ha determinado que existen dos genes asociados con la aparición de THH, estos genes se han visto que codifican para proteínas de trans membrana de las células endoteliales que son componentes de la superfamilia de factores decrecimiento transformante beta, y se produce una alteración entre la proteína ósea morfogénica 9 que actúa como antiangiogénico y el factor de crecimiento del endotelio vascular, proangiogénico. Debido a esto, se ha considerado el uso de bevacizumab que inhibe la angiogénesis y el factor de crecimiento del endotelio vascular (12-14).
Existen varios reportes de casos y serie de casos con el uso de bevacizumab como terapia local aplicado de forma intramucosas en región nasal con disminución de sangrados nasales, y de aplicación en forma sistémica con disminución de sangrado, soporte transfusional, y en algunos casos mejoría de manifestaciones sistémicas de la enfermedad con disminución de la falla cardiaca de alto gasto (15-19).
En un estudio fase II en pacientes con diagnóstico THH con malformaciones arteriovenosas hepáticas y falla cardiaca de alto flujo se evaluó el uso de bevacizumab a una dosis de 5mg/kg cada dos semanas por seis aplicaciones, el objetivo principal del estudio era evaluar la función cardiaca con disminución de la fracción de eyección medida a los tres meses de iniciado el tratamiento. Ingresaron al estudio 24 pacientes todos con malformaciones arteriovenosas hepáticas y falla cardiaca de alto gasto con índice cardiaco de antes de iniciar el tratamiento de 5.01 L/min/m2, se encontró respuesta al tratamiento en 20 de los 24 pacientes, con normalización del índice cardiaco en tres, con respuesta parcial en 17 y en cuatro pacientes sin respuesta. Como hallazgos adicionales se halló una disminución de la epistaxis en 87%de los pacientes, mejoría en los niveles de hemoglobina y disminución en la necesidad de transfusión, sin cambios imagenológicos en las malformaciones arteriovenosas, ni en las telangiectasias (20).
La paciente reportada en este momento con THH, con gran compromiso de malformaciones vasculares viscerales, tanto hepáticas como pulmonares, que la han llevado a una falla cardiaca de alto flujo e hipertensión pulmonar severa, con anemia dependiente de soporte transfusional, se consideró el uso de bevacizumab ante falla de tratamientos previos.
Se encontró muy buena respuesta clínica, disminución progresiva del sangrado con recuperación de la anemia, tornándose independiente de soporte transfusional y disminución de los edemas, con mejor tolerancia al ejercicio e independencia de suplemento del oxígeno.
Sin embargo, la respuesta clínica fue transitoria, estuvo seis meses libre de tratamiento y transfusiones, retornando nuevamente a sus episodios descritos previamente, lo cual está escrito en la literatura; pero con buena respuesta al reinicio del manejo con normalización de niveles de hemoglobina y de la clase funcional, considerando este medicamento una opción adecuada para el uso en esta rara enfermedad cuando existen manifestaciones sistémicas severas.
Por el momento desconocemos la dosis sistémica de bevacizumab para el manejo de manifestaciones severas de la enfermedad y el tiempo de duración, por el cual se debe recibir el medicamento, ya que al suspenderlo existe reaparición en la sintomatología de la enfermedad.
Referencias
1. Dheyauldeen S, Abdelnoor M, Bachmann-Harildstad G. The natural history ofepistaxis in patients with hereditary hemorrhagic telangiectasia in the Norwegian population: a cross-sectional study. Am J Rhinol Allergy. 2011 Jul-Aug; 25(4): 214-8. [ Links ]
2. Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, Kjeldsen AD, Plauchu H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000; 91(1): 66. [ Links ]
3. Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, Spears J, Brown DH, Buscarini E, Chesnutt MS, Cottin V, Ganguly A, Gossage JR, Guttmacher AE, Hyland RH, Kennedy SJ, Korzenik J, Mager JJ, Ozanne AP, Piccirillo JF, Picus D, Plauchu H, Porteous ME, Pyeritz RE, Ross DA, Sabba C, Swanson K, Terry P, Wallace MC, Westermann CJ, White RI, Young LH, Zarrabeitia R, HHT Foundation International - Guidelines Working Group. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia J Med Genet. 2011; 48(2): 73. [ Links ]
4. Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997; 390(6659): 465. [ Links ]
5. Sadick H, Riedel F, Naim R, Goessler U, Hörmann K, Hafner M, Lux A. Patients with hereditary hemorrhagic telangiectasia have increased plasma levels of vascular endothelial growth factor and transforming growth factor-beta1 as well as high ALK1 tissue expression. Haematologica. 2005; 90(6): 818. [ Links ]
6. Hsu CC, Kwan GN, Thompson SA, Evans-Barns H, van Driel ML. Embolisation for pulmonary arteriovenous malformation. Cochrane Database Syst Rev. 2012; 8: CD008017. [ Links ]
7. Lund VJ, Howard DJ. A treatment algorithm for the management of epistaxis in hereditary hemorrhagic telangiectasia. Am J Rhinol. 1999; 13(4): 319. [ Links ]
8. Hitchings AE, Lennox PA, Lund VJ, Howard DJ. The effect of treatment for epistaxis secondary to hereditary hemorrhagic telangiectasia. Am J Rhinol. 2005;19(1): 75. [ Links ]
9. Bergler W, Sadick H, Gotte K, Riedel F, Hörmann K. TI Topical estrogens combined with argon plasma coagulation in the management of epistaxis in hereditary hemorrhagic telangiectasia. Ann Otol Rhinol Laryngol. 2002; 111: 222. [ Links ]
10. Folz BJ, Tennie J, Lippert BM, Werner JA. Natural history and control of epistaxis in a group of German patients with Rendu-Osler-Weber disease. Rhinology. 2005; 43(1): 40. [ Links ]
11. Buscarini E, Leandro G, Conte D, Danesino C, Daina E, Manfredi G, Lupinacci G, Brambilla G, Menozzi F, De Grazia F, Gazzaniga P, Inama G, Bonardi R, Blotta P, Forner P, Olivieri C, Perna A, Grosso M, Pongiglione G, Boccardi E, Pagella F, Rossi G, Zambelli A. Natural history and outcomeof hepatic vascular malformations in a large cohort of patients with hereditary hemorrhagic teleangiectasia. Dig Dis Sci. 2011 Jul; 56(7): 2166-78. [ Links ]
12. Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009; 17(7): 860. [ Links ]
13. Berg JN, Gallione CJ, Stenzel TT, Johnson DW, Allen WP, Schwartz CE, Jackson CE, Porteous ME, Marchuk DA. The activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet. 1997; 61(1): 60. [ Links ]
14. McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J. Endoglin, a TGFbeta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994; 8(4): 345. [ Links ]
15. Simonds J, Miller F, Mandel J, Davidson TM The effect of bevacizumab (Avastin) treatment on epistaxis in hereditary hemorrhagic telangiectasia. Laryngoscope.2009; 119(5): 988 [ Links ]
16. Bose P, Holter JL, Selby GB. Bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2009; 360(20): 2143. [ Links ]
17. Oosting S, Nagengast W, de Vries E. More on bevacizumab in hereditary hemorrhagic telangiectasia. N Engl J Med. 2009; 361(9): 931; author reply 931. [ Links ]
18. Flieger D, Hainke S, Fischbach W. Dramatic improvement in hereditary hemorrhagic telangiectasia after treatment with the vascular endothelial growth factor (VEGF) antagonist bevacizumab. Ann Hematol. 2006;85(9):631 [ Links ]
19. Mitchell A, Adams LA, MacQuillan G, Tibballs J, vanden Driesen R, Delriviere L. Bevacizumab reverses need for liver transplantation in hereditary hemorrhagic telangiectasia. Liver Transpl. 2008; 14(2): 210. [ Links ]
20. Dupuis-Girod S, Ginon I, Saurin JC, Marion D. Bevacizumab in Patients With Hereditary Hemorrhagic Telangiectasia and Severe Hepatic Vascular Malformations and High Cardiac Output. JAMA. 2012; 307(9): 948-955 [ Links ]
