










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Medica Colombiana
Print version ISSN 0120-2448
Acta Med Colomb vol.40 no.3 Bogotá July/Dec. 2015
Presentación de casos
Apoplejía tumoral pituitaria
Pituitary tumor apoplexy
Adrián Zamora, Paola Martínez, Hernán Bayona
1 Residente de Cuarto Año de Neurología, Universidad El Bosque-Fundación Santa Fe de Bogotá; Bogotá, D.C. (Colombia). E-mail: adrianzamora707@gmail.com
2 Dra. Paola Martínez: Interna Universidad de los Andes - Fundación Santa Fe de Bogotá; Bogotá, D.C. (Colombia).
3 Neurólogo Fundación Santa Fe de Bogotá. Epidemiólogo Clínico. Bogotá, D.C. (Colombia).
Recibido: 03/I/2015. Aceptado: 31/VIII/2015
Resumen
La apoplejía tumoral pituitaria es un síndrome infrecuente que resulta del infarto y/o hemorragia espontánea de un adenoma pituitario preexistente. Ya que el evento primario involucra el adenoma, este síndrome debe ser nombrado como apoplejía tumoral pituitaria y no como apoplejía pituitaria. El aumento súbito en la presión de los contenidos de la silla turca da como resultado una cefalea de inicio agudo (puede ser incluso una “cefalea en trueno”) de intensidad severa, alteraciones visuales y compromiso en la función pituitaria. El diagnóstico se basa en una alta sospecha clínica, imagen por resonancia magnética y medición de hormonas hipofisiarias en sangre. El tratamiento se basa en medidas de soporte (líquidos intravenosos y corticoides) y en casos sin buena respuesta o con deterioro neurológico, descompresión de silla turca. A continuación presentamos el caso de un adenoma previamente no diagnosticado que debutó como apolejía tumoral pituitaria. (Acta Med Colomb 2015; 40: 249-253).
Palabras clave: Apoplejía pituitaria, adenoma pituitario, cefalea en trueno, diplopía, hemorragia pituitaria, hipopituitarismo, insuficiencia adrenal, leuprolida, oftalmoplejía dolorosa.
Abstract
Pituitary tumor apoplexy is an infrequent condition resulting from infarction and/or spontaneous bleeding from a pre-existing pituitary adenoma. This entity requires the prior existence of an adenoma in order to be named as pituitary tumor apoplexy, otherwise, it should be named pituitary apoplexy. The sudden increase in pressure of the sella turcica's contents results in a clinical syndrome characterized by headache (which can be “thunderclap headache”), visual disturbances and hypopituitarism. Diagnosis is not always straight forward and requires high clinical suspicion in addition to magnetic resonance imaging and measurement of serum pituitary hormones. Treatment is mainly based on supportive measures (intravenous fluids and steroids) and surgical decompression in those cases with no response to medical treatment and progressive neurological impairment. We report the case of a patient with a previously unknown pituitary adenoma presenting as a tumor apoplexy. (Acta Med Colomb 2015; 40: 249-253).
Keywords: pituitary apoplexy, pituitary adenoma, thunderclap headache, diplopia, pituitary hemorrhage, hypopituitarism, adrenal insufficiency, leuprolide, painful ophthalmoplegia.
Introducción
La apoplejía tumoral pituitaria es un síndrome poco frecuente que se presenta por hemorragia o infarto espontáneo de un adenoma pituitario preexistente (1). Debido a que el evento primario implica la preexistencia de un adenoma, el síndrome debe ser referido como apoplejía tumoral pituitaria y no como apoplejía pituitaria (1,2). El aumento súbito de la presión intraselar y de sus contenidos resulta en un síndrome clínico caracterizado por cefalea (en algunos casos cefalea en trueno), alteraciones visuales y deterioro de la función pituitaria. El manejo inicial de los pacientes incluye tratamiento de soporte (líquidos endovenosos y corticoides), después de lo cual muchos pacientes presentan una mejoría clínica. Los casos más severos o con deterioro clínico podrían beneficiarse de una descompresión quirúrgica urgente por un neurocirujano experimentado (1). Todos los pacientes que se presentan con este síndrome requieren a largo plazo un seguimiento de la función hipofisiaria e identificación y tratamiento temprano de cualquier tumor residual.
Presentación del caso
Se trata de un paciente de 63 años de edad procedente de Bogotá, quien acude al servicio de urgencias por cuadro de siete días de cefalea hemicránea y retroocular izquierda de intensidad 7/10. El dolor inició en forma súbita, después de realizar actividad física (trotar) y alcanzó su máxima intensidad en segundos. No se asoció a pérdida de conciencia o vómito. Acudió al servicio de urgencias donde se hizo una TAC de cráneo que fue reportada inicialmente como normal (Figura 1). En esa ocasión se dio de alta con antiinflamatorios no esteroideos con los que hubo una mejoría parcial del cuadro; sin embargo, 72 horas después el paciente reingresa por persistencia del dolor asociado a diplopía horizontal que empeoraba al mirar hacia la izquierda y astenia. En la revisión por sistemas, manifestaba pérdida de la libido de varios meses de evolución. Como antecedentes el paciente tuvo un cáncer de próstata con resección total en 2001,con una recaída en 2013 que requirió radioterapia y terapia antiandrogénica con leuprolida (agente agonista de GnRH).
Al examen físico de ingreso FC: 78/min, FR: 19/min, TA134/70 mmHg, con un examen físico general normal. Al examen neurooftalmológico el fondo de ojo sin edema de papila, la agudeza visual 20/25 bilateral con pupilas simétricas de 3 mm reactivas al estímulo fotomotor directo y consensual; en la mirada en reposo se evidenciaba exotropia izquierda con movimiento de refijación del ojo izquierdo al realizar el test de oclusión alternante. Al examinar los movimientos oculares se encontró paresia del recto medial izquierdo y ptosis del mismo lado. El resto del examen neurológico se encontraba dentro de límites normales; en especial no había compromiso de otros pares craneanos, la fuerza y los reflejos estaban conservados. Se tomaron unos laboratorios básicos de ingreso en los que se encontró un sodio plasmático bajo 126.3 mEq/L. El resto de exámenes (hemograma, BUN, creatinina, y K) fueron normales.
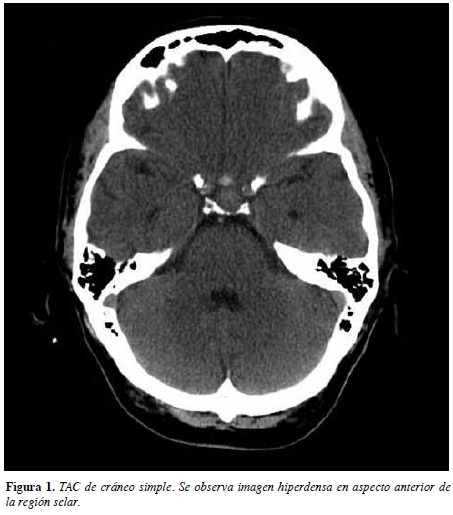
Con los datos anteriores se consideró un diagnóstico inicial de cefalea en trueno, oftalmoplejía dolorosa e hiponatremia a estudio. Se sospechó patología de región selar o del seno cavernoso y se revisó nuevamente la TAC anterior en la que evidenció una imagen hiperdensa en el aspecto anterior de la región selar sugestiva de patología pituitaria (Figura 1). Se solicitó una resonancia magnética de cerebro con medio de contraste en la que se evidenció una masa selar de 16 milímetros de diámetro con captación del medio de contraste y señal heterogénea en su interior (hiperintensa en T1 e hipointensa en T2), correspondiente a un macroadenoma hipofisiario con sangrado subagudo (Figuras 2 y 3).
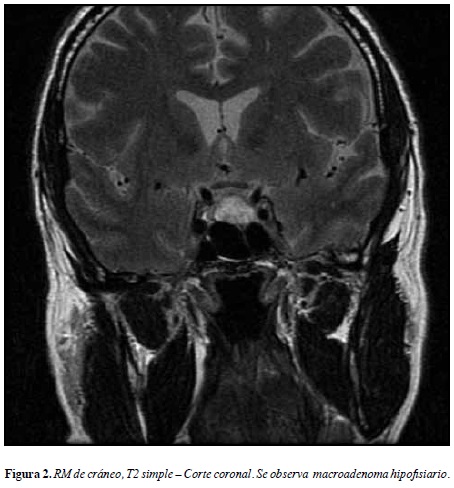
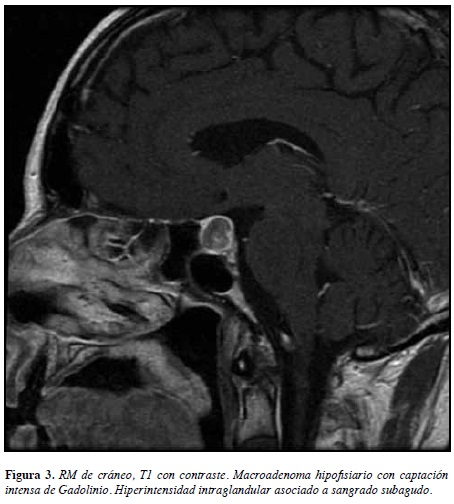
Teniendo en cuenta los hallazgos radiológicos y clínicos, se hizo un diagnóstico de apoplejía tumoral pituitaria con transformación hemorrágica. Se solicitó un estudio hormonal que reportó testosterona en 0.0 ng/dL y cortisol disminuido con unos niveles de 30.6 ug/L (60-250 ug/L). Los niveles de hormona adrenocorticotrópica, las pruebas tiroideas, la prolactina, la hormona de crecimiento y el factor I de crecimiento similar a la insulina fueron normales.
Se inició administración de líquidos endovenosos e hidrocortisona 100 mg IV bolo seguido de 50 mg IV cada 12 horas. El paciente fue valorado en conjunto con los servicios de neurocirugía y endocrinología y dada la estabilidad hemodinámica y neurológica del paciente, se consideró continuar manejo médico de soporte y no llevar a descompresión urgente de silla turca. El paciente continuó recibiendo suplencia con corticoides y en corrección de hiponatremia y a los siete días fue llevado a resección transesfenoidal encontrándose un hematoma antiguo coagulado en región selar, con un tumor de aspecto benigno, que fue extraído, dejando indemne el lóbulo posterior de la hipófisis.
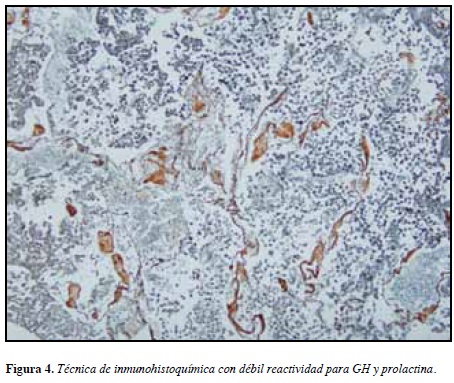
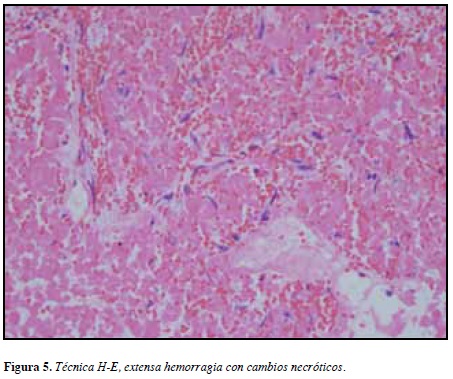
Los hallazgos de patología en estudios de histoquímica y estudios de inmunohistoquímica evidenciaron necrosis isquémica y cambios compatibles con apoplejía de un adenoma pituitario (Figuras 4 y 5), con débil reactividad para la GH y prolactina; sin embargo, dadas las características necróticas del tumor no se pudo establecer con certeza su naturaleza.
Desde el egreso el paciente ha venido evolucionando en forma satisfactoria, con mejoría completa de la oftalmoplejía y normalización de la función hormonal con suplencia de corticoides y testosterona.
Discusión
El primer caso de apoplejía pituitaria fue descrito en un paciente con acromegalia por Pearce Bailey en 1898, y en 1095, Bleibtreu describió una serie de pacientes con tumores pituitarios que se presentaron con cefalea súbita, vómito, disminución de la agudeza visual y disfunción hormonal. Casi medio siglo después, en 1950, Brougham et al estandarizaron el síndrome clínico en una serie de casos cuyo desenlace era la muerte en todos los pacientes (1).
Es importante diferenciar esta entidad de la apoplejía no tumoral pituitaria, la cual se presenta en una glándula libre de tumor, como el clásico síndrome de Sheehan por hemorragia posparto, en casos de apoplejía secundaria a shock séptico (2) o ruptura de un quiste de Rathke (3). Hay que considerar que hasta en 25% no producen síntomas (4) y no requieren el enfoque terapéutico de urgencia de una apoplejía clínicamente manifiesta.
A pesar de ser una entidad descrita hace más de 50 años, su diagnóstico aún sigue siendo desafiante por su presentación clínica heterogénea y porque la mayoría (80%) de los pacientes no tienen diagnosticado el adenoma al momento del cuadro (1, 5-7). En el caso presentado, la asociación de cefalea con síntomas neurooftalmológicos e hiponatremia generó la sospecha diagnóstica (1). Una revisión de casos reciente estableció que el síntoma más frecuente y precoz es la cefalea súbita de localización retroorbitaria seguida de la disminución de la agudeza visual y la diplopía, las cuales se presentan hasta en 70% de los casos (6, 7). Es importante realizar un diagnóstico diferencial con otras causas de cefalea de inicio súbito y oftalmoplejía dolorosa como la hemorragia subaracnoidea y la trombosis del seno cavernoso(1, 5, 14). Habitualmente este diagnóstico diferencial se realiza mediante resonancia de cerebro contrastada y angiotac.
El hipopituitarismo es la principal causa de morbilidad y mortalidad de la apoplejía tumoral pituitaria. El déficit más importante es el de ACTH, ya que puede desencadenar una insuficiencia suprarrenal aguda fatal (1, 7, 10). Otro déficit frecuente (70%) es la insuficiencia gonadal (6, 7), por lo que es importante preguntar al paciente sobre pérdida de la libido, cómo en el caso presentado. Llama la atención que la insuficiencia suprarrenal aguda fue predominantemente subclínica y que en ningún momento se acompañó de inestabilidad hemodinámica, hipoglicemia o hipercalemia, lo cual puede suceder hasta en 30% de los casos según series recientes (6, 7, 12, 14). El único síntoma sugestivo de insuficiencia suprarrenal fue la debilidad, aunque ésta también pudo asociarse a la hiponatremia, la cual en casos de apoplejía pituitaria es habitualmente producida por un síndrome de secreción inapropiada de hormona antidiurética (SIADH).
En la mayoría de los casos la apoplejía tumoral pituitaria ocurre en forma espontánea; sin embargo, en 25-30% de los pacientes se han identificado factores precipitantes como intervenciones quirúrgicas, la estimulación de la glándula en pruebas funcionales y la anticoagulación, entre otros (9). En el caso presentado se identificó como probable desencadenante el uso previo de leuprolida, un agonista de GnRH usado en cáncer de próstata que en algunos reportes de caso ha sido asociado con apoplejía tumoral pituitaria (24,25). Sin embargo, es difícil establecer una relación causal clara debido al desconocimiento del tiempo de evolución del adenoma y a la falta de evidencia que establezca una asociación causal significativa entre el uso de este medicamento y el infarto de un tumor pituitario.
Con respecto al uso de neuroimágenes, la tomografía axial computarizada es con frecuencia la primera imagen que se le practica a los pacientes, y si bien su sensibilidades baja en el contexto de una apoplejía pituitaria (46%), no debería ser subestimada ya que puede identificar la transformación hemorrágica de un infarto pituitario y llevar a un diagnóstico precoz en algunos casos (13). Sin embargo, para un diagnóstico certero el estándar de oro sigue siendo la resonancia magnética con contraste y secuencia dinámica de hipófisis, con una sensibilidad y especificidad cercanasa 95%.
Los pilares del tratamiento de esta patología son el monitoreo del paciente, la administración de líquidos endovenosos, corticoides y en algunos casos la descompresión quirúrgica. Tradicionalmente se ha considerado la apoplejía tumoral pituitaria como una urgencia neuroquirúrgica; sinembargo, no todos los casos lo son. La decisión de intervenir inmediatamente o diferir la cirugía (como en el caso presentado) debe ser tomada en el contexto de cada paciente, siendo el manejo conservador una opción viable en aquellos pacientes con microadenomas, síntomas mínimos, buen estatus neurológico, sin compromiso visual y buena respuesta a los corticoides (17-19, 22). Con respecto al tiempo de inicio de corticoides, éste depende de la estabilidad hemodinámica, del estado de conciencia y de la agudeza visual. Si el paciente se encuentra estable hemodinámicamente, sin alteración del estado de conciencia ni disminución de la agudeza visual, se recomienda diferir el inicio empírico de los mismos (17) hasta tomar una muestra de cortisol a las 9 am y en caso de ser ésta < 550 nmol/L, iniciar suplencia inmediata. De lo contrario, se debe iniciar de forma empírica hidrocortisona 100-200 mg IV en bolo y seguir con 50 mgIV de cada seis horas (1, 6, 17, 22).
En cuanto al pronóstico de esta entidad, se observa una mejoría completa de la agudeza visual y los déficit hormonales en la mayoría de los pacientes (70%). Sinembargo, su pronóstico sigue siendo malo en los casos no identificados ni tratados en forma precoz, alcanzando una mortalidad cercana al 80%. (1, 6, 7, 10).
Agradecimientos Al Departamento de Neurología de la Fundación Santa Fe. Al paciente, quien en forma estrechamente gentil permitió la publicación de este caso. Al Dr. Nicolás Useche del Departamento de Neuroradiología y al Dr. Rafael Andrade de Patología quienes facilitaron las imágenes y los estudios de patología.
Fuentes de financiación y conflictos de interés
Este caso no tiene financiación ni conflictos de interés.
Referencias
1. Edward R. Laws. Pituitary Tumor Apoplexy: A Review. J Inten Care Med Volume 23 Number 2 March/April 2008 75-90. [ Links ]
2. Bahauddin M. Partial hypopituitarism following septic peritonitis with shock. Arch. Intern. Med. 1978; Aug; 138 (8): 1272-3. [ Links ]
3. Kleinschmidt-DeMasters BK, Lillehei KO, Stears JC. The pathological, surgical and MR spectrum of Rathke cleft cysts. Surg Neurol 1995; 44: 19-27. [ Links ]
4. Wakai S, Fukushima T, Teramoto A, et al. Pituitary apoplexy: Its incidence and clinical significance. J Neurosurg 1981; 55: 187-193. [ Links ]
5. Nielsen EH, Lindholm J, Bjerre P et al. Frequent ocurrence of pituitary apoplexyin patients with non-functioning pituitary adenoma. Clin Endocrinol (Oxf). 2006;64: 319-322. [ Links ]
6. Verrees M et al. Pituitary tumor apoplexy: characteristics, treatment and outcomes. Neurosurg Focus. 2004; 16: 1-7. [ Links ]
7. Mehmet Turgut, Yelda Özsunar, Sema Başak, Engin Güney, Erkin Kır, İbrahim Meteoğlu. Pituitary apoplexy: an overview of 186 cases published during the last century. Acta Neurochir 2010; 152: 749-761. [ Links ]
8. Kruse et al. Pressure and blood flow in pituitary adenomas measured during transsphenoidal surgery. Br J Neurosurg. 1992; 6: 333-341. [ Links ]
9. Patrick L. Semple, John A. Jane, Jr., Edward R. Laws, Jr. Clinical Relevance of Precipitating Factors in Pituitary Apoplexy. Neurosurgery 2007; 61: 956-962. [ Links ]
10. Latika Sibal, Steve G. Ball, Vincent Connolly, Robert A. James, Philip Kane, William F. Kelly, Pat Kendall-Taylor, David Mathias, PetrosPerros, Richard Quinton, Bijay Vaidy. Pituitary Apoplexy: A Review of Clinical Presentation, Management and Outcome in 45 Cases. Pituitary, 2004; 7: 157-163. [ Links ]
11. Arafah BM. Reversible hypopituitarism in patients with large non-functioning pituitary adenomas. J Clin Endocrinol Metab. 1986; 62: 1173-1179. [ Links ]
12. Veldhuis JD, Hammond JM. Endocrine function after spontaneous infarction ofthe human pituitary: report, review, and reappraisal. Endocr Rev. 1980; 1: 100-107. [ Links ]
13. Post MJ, David NJ, Glaser JS, Safran A. Pituitary apoplexy: diagnosis by computed tomography. Radiology. 1980; 134: 665-670. [ Links ]
14. Chibbaro S, Benvenuti L, Crnesecchi S, Faggionato F, Gagliardi R. An interesting case of a pituitary adenoma apoplexy mimicking an acute meningitis. Case report. J Neurosurg. 2007 Jun;51 (2): 65-9; discussion 68-9. [ Links ]
15. Lazaro CM, Guo WY, Sami M, et al. Hemorrhagic pituitary tumours. Neuro radiology. 1994; 36: 111-11. [ Links ]
16. Lucas J, Zada G. Imaging of the pituitary and parasellar region. Semina neuro,2012; 32: 320-331. [ Links ]
17. UK Guidelines for the management of Pituitary Apoplexy. Clinical Endocrinology 2011; 74: 9-20. [ Links ]
18. Maccagnan P, Macedo CL, Kayath MJ, Nogueira RG, Abucham J. Conservative management of pituitary apoplexy: a prospective study. J Clin Endocrinol-Metab. 1995; 80: 2190-2197. [ Links ]
19. Ayuk J. Acute management of pituitary apoplexy surgery or conservative management. Clin Endocrinol (Oxf). 2004; 61: 747-752. [ Links ]
20. Zayour DH, Selman WR, Arafah BM. Extreme elevation of intrasellar pressure in patients with pituitary tumor apoplexy: relation to pituitary function. J Clin Endocrinol Metab. 2004; 89: 5649-5654. [ Links ]
21. Arafah BM, Harrington JF, Madhoun ZT, Selman WR. Improvement of pituitary function after surgical decompression for pituitary tumor apoplexy. J Clin EndocrinolMetab. 1990; 71: 323-328. [ Links ]
22. Vicente A, Lecumberri A, Gálvez B. Clinical practice guideline for the diagnosis and treatment of pituitary apoplexy. Endocrinol Nutr 2013; 60 (10): 582. e1-582. e12. [ Links ]
23. Muthukumar, Rosette N, Soundaram M. Blindness following pituitary apoplexy: Timing of surgery and neuro-ophthalmic outcome. J Clin Neurosci. 2008; 15: 873 - 879. [ Links ]
24. Guerra Y, Lacuesta E, Marquez F et al. Apoplexy in non functioning pituitary adenoma after one dose of leuprolide as treatment for prostate cancer. Pituitary2010; 13 (1): 54-59. [ Links ]
25. Davis A, Goel S, Picolos M, Wang M, Lavis V. Pituitary apoplexy after leuprolide. Pituitary 2006; 9 (3): 263-265. [ Links ]
