










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActa Medica Colombiana
versión impresa ISSN 0120-2448
Acta Med Colomb vol.40 no.4 Bogotá oct./dic. 2015
Presentación de Casos
Síndrome de Löfgren
Una variante clínica de la sarcoidosis caracterizada por eritema nodoso, artritis y adenomegalias mediastinales
Löfgren's syndrome
A clinical variant of sarcoidosis characterized by erythema nodosum, arthritis, and mediastinal lymphadenopathy
Diego Severiche-Hernández, Diego Fernando Severiche-Bueno, Débora Rey •
Bogotá, D.C. (Colombia)
Dr. Diego Severiche-Hernández: FACCP. FACP. Internista. Neumólogo. Intensivista. Especialista en Bioética. Especialista en Educación Médica. Docente Área de Medicina Interna. Facultad de Medicina Universidad de La Sabana. Neumólogo. Clínica del Country. Clínica Shaio. Intensivista UCI Hospital San Rafael Tunja;
Dr. Diego Fernando Severiche-Bueno: Médico Interno. Universidad de La Sabana;
Dra. Débora Rey O: Universidad de La Sabana. Bogotá, D.C. (Colombia).
Correspondencia. Dr. Diego Fernando Severiche. Bogotá, D.C. (Colombia).
E-mail: diego.severiche1@unisabana.edu.co
Recibido: 02/I/2015 Aceptado: 21/X/2015
Resumen
Se presenta el caso de una paciente de 24 años que consulta por inflamación de los tobillos y eritema nodoso. Al realizar el estudio clínico y paraclínicos de la paciente se diagnosticó síndrome de Löfgren, entidad de baja prevalencia en nuestro país. Este caso permite realizar una discusión del diagnóstico del eritema nodoso y la sarcoidosis. (Acta Med Colomb 2015; 40: 345-348).
Palabras clave: síndrome de Löfgren, eritema nodoso, sarcoidosis, granulomas, artralgia.
Abstract
The case of a patient of 24 years who consulted for swollen ankles and erythema nodosum is presented. In conducting clinical and laboratory study of patient, Löfgren's syndrome diagnosis was made, entity of low prevalence in our country. This case allows to make a discussion of the diagnosis of erythema nodosum and sarcoidosis. (Acta Med Colomb 2015; 40: 345-348).
Keywords: Lofgren's syndrome, erythema nodosum, sarcoidosis, granulomas, arthralgia.Introducción
La sarcoidosis es una enfermedad sistémica de causa desconocida, que es caracterizada por la formación de granulomas inmunes en varios órganos, principalmente el pulmón y ganglios linfáticos (1-3). Estudios muestran que la sarcoidosis puede ser el resultado de una reacción granulomatosa exagerada después de la exposición a antígenos no identificados en individuos que son genéticamente susceptibles (2, 4, 5). El síndrome de Löfgren es una forma de la presentación aguda de la sarcoidosis (6). Se presenta un caso de síndrome de Löfgren, entidad de baja prevalencia en nuestro medio, pero de alto interés académico.
Caso clínico
Paciente femenino de 24 años de edad, quien consulta al servicio de urgencias por presentar cuadro clínico de 12 horas de evolución consistente en edema maleolar bilateral doloroso a la palpación y a la movilización. Asociado al cuadro clínico la paciente refiere rash eritematoso en la región superior del maléolo interno de la pierna izquierda y rodillas. La paciente niega cualquier tipo de antecedente patológico, quirúrgico, toxicológico y farmacológico.
Al examen físico se encuentra paciente, normotérmica, normotensa, orientada en tiempo y espacio. Como hallazgos positivos se evidencia calor, rubor y edema maleolar bilateral sin fóvea doloroso a la palpación (Figura 1). Se realiza cuadro hemático, parcial de orina, BUN, creatinina y proteína C reactiva los cuales se encontraban dentro de límites normales. Le dan salida con AINEs, incapacidad médica y control por medicina interna.
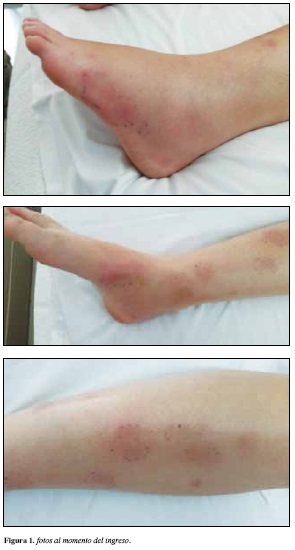
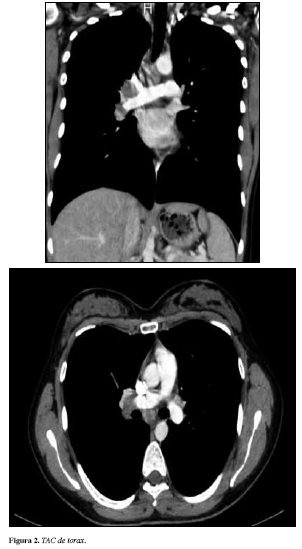
La paciente manifiesta que el edema y el dolor maleolar se incrementan al punto que le impide movilizarse, adicionalmente refiere fiebre cuantificada en 39°C asociada a la aparición del rash eritematoso en miembros inferiores, doloroso a la palpación. Cuadro que persiste alrededor de 10 días. Valorada ambulatoriamente por medicina interna, se evidencia lesiones en piel compatibles con eritema nodoso, e inflamación articular a nivel de tobillos bilateralmente. El resto del examen físico de la paciente se encontró dentro de límites normales. El cuadro era incapacitante, razón por la cual se decide hospitalización para estudio y manejo de dolor por limitación funcional. Los paraclínicos evidencian cuadro hemático con leucocitosis leve sin anemia; VSG ligeramente aumentada. Función renal normal. Parcial de orina: normal. Factor reumatoide negativo; PCR en 56.1; anticuerpos ANA negativos, anticuerpos anti DNA negativo y C3 y C4 normal. Se mide HLA DR3 el cual es positivo. Dentro de las imágenes diagnosticas se toma una radiografía de pies que evidencia relaciones articulares normales, con edema de tejidos blandos periarticulares. Se realiza TAC de tórax (Figura 2) que muestra ganglios aumentados de tamaño hasta de 18 mm en estaciones hiliar derecha, subcarinal y precarinal. Parénquima pulmonar normal sin presencia de derrames pleurales.
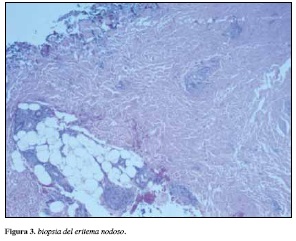
Posteriormente se le realizó biopsia de piel de pierna derecha reportada como paniculitis septal con infiltrado con linfocitos, histiocitos, polimorfonucleares y granulomas con células gigantes multinucleadas (Figura 3) hallazgos compatibles con eritema nodoso.
A la paciente se le inicio manejo con AINEs, y terapia física sedativa, con pobre respuesta persistiendo dolor articular, fiebre y lesiones eritematosas, razón por la cual se decidió iniciar un curso corto de corticoides orales, a dosis antiinflamatoria, con lo cual se logra una respuesta inmediata y favorable. En el tratamiento intrahospitalario participaron los especialistas de medicina interna, reumatología y dermatología de la institución. La paciente evolucionó favorablemente con desaparición de los signos y síntomas y; sin necesidad de tratamiento farmacológico ambulatorio.
Discusión
El eritema nodoso se considera una paniculitis que afecta la grasa subcutánea de la piel (7). Es más frecuente en mujeres y tiene un pico de incidencia entre los 18 y 34 años (8). La incidencia anual está entre 1 a 5 por 100 000 personas (7). Entre el 17 y 72% de los casos son idiopáticos (8). El exacto mecanismo del eritema nodoso no está completamente establecido, pero se considera puede ser el resultado del depósito de complejos inmunes en las vénulas de los septos de la grasa subcutánea causando así una paniculitis neutrofílica (8, 9). Es posible que exista una predisposición genética (8). Estos aspectos tienen similitud con la fisiopatología de la sarcoidosis, como se verá más adelante.
Característicamente las lesiones cutáneas son nódulos rojos, simétricos, dolorosos, localizados más frecuentemente en la cara anterior de las piernas, aunque se pueden presentar en cualquier parte del cuerpo y varían de 1 a 5 cm de diámetro. Involucionan en el curso de días o semanas, con la apariencia de pequeñas contusiones, no se ulceran y tienden a mejorar completamente (7-9).
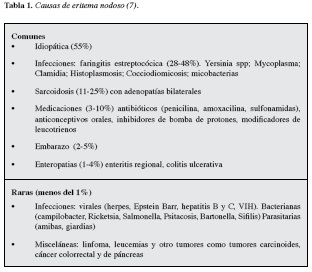
Las causas del eritema nodoso se describen en la Tabla 1, la cual nos permite ver que aunque el porcentaje es relativamente bajo (11-25%), la sarcoidosis es una de las principales causas por considerar en el momento de abordar el eritema nodoso (7).
La sarcoidosis es una enfermedad que ha sido reconocida desde hace más de 120 años, y aun hoy sigue siendo una enfermedad con muchas áreas grises (10). Es una entidad presente en todo el mundo, cuya prevalencia es más alta en Europa y en Estados Unidos. Es más predominante en personas afroamericanas, particularmente en mujeres (10).
En Colombia, la prevalencia es baja aunque no está claramente establecida. Una revisión, realizada por el doctor Valovis en 1977 reportó 51 casos en la ciudad de Bogotá en donde sólo un caso se presentó como síndrome de Löfgren. (11). La exacta causa de la sarcoidosis es desconocida. Muchos estudios sugieren una susceptibilidad genética así como factores ambientales que pueden contribuir al desarrollo de la enfermedad. Inmunológicamente, la sarcoidosis es una respuesta inmune exagerada a antígenos hasta ahora no identificados (10, 12-14). Esto lo podemos extrapolar con el caso donde se observó que la paciente no tenía antecedentes de importancia que pudieran ayudar en el abordaje de su cuadro clínico.
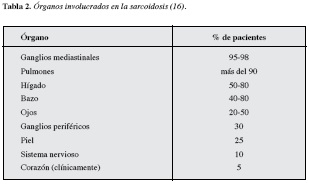
No hay evidencia que la sarcoidosis sea una enfermedad infecciosa. En tanto, sí hay una respuesta inmune exagerada a patrones moleculares asociados a patógenos de micobacterias y propionibacterias muertas o parcialmente degradadas que promueve la agregación y persistencia de antígenos no degradables, formando un nido para la formación de granulomas. Esta formación causa lesiones sarcoides a través de una respuesta inmune exagerada debido a una interrelación entre macrófagos y las células T que se estimulan mutuamente (10, 15). También se han descrito otras sustancias orgánicas e inorgánicas que pueden desencadenar la sarcoidosis (10). Los órganos que principalmente afectan la sarcoidosis se resumen en la Tabla 2 dejando ver que en el caso clínico se presentó el compromiso ganglionar que es lo más común pero no presentó compromiso en otros órganos del cuerpo aparte de la piel que sólo se presenta en el 25% de los pacientes (12).
Los principios básicos para el diagnóstico son: la clínica con la presentación radiológica; la evidencia de granulomas no caseificantes; y la evidencia de ausencia de enfermedades alternativas (18). Los criterios diagnósticos se resumen en la Tabla 3.

Con respecto al síndrome de Löfgren, este fue descrito por primera vez por Löfgren y Lundback en 1952 (1). Este síndrome se ha considerado como una presentación aguda de la sarcoidosis, caracterizado por la presencia de la triada de eritema nodoso, adenomegalias hiliares bilaterales, artritis o artralgias que característicamente compromete los tobillos (2, 3, 17). Un 35% de los casos de sarcoidosis pueden comenzar como un síndrome de Löfgren. Es más común en mujeres jóvenes de raza blanca, y es más rara en personas de raza negra lo cual se correlaciona con el caso expuesto (2).
El síndrome se puede acompañar de otros síntomas y signos como fiebre (38%), tos y disnea (13%), hepatomegalia (6%), esplenomegalia (2%), síntomas oculares (5%), adenomegalias periféricas (4%), hipercalcemia (2%), hipertrofia de glándulas salivales (1%) o compromiso de SNC(1%) (2, 3). También se ha descrito la presencia de miopatía y compromiso peritoneal (3, 18). En el caso expuesto la paciente no presentó ninguna de las otras manifestaciones mencionadas.
El diagnóstico de síndrome de Löfgren requiere la presencia de dos de los tres síntomas cardinales; la artritis o artralgias pueden o no estar presentes (2). Como en nuestro caso; se ha demostrado la asociación con el HLA DRB1*03 (4, 5). Su presencia se relaciona con un curso favorable de la enfermedad, con un pronóstico excelente, y la remisión de los síntomas en los primeros dos años de la enfermedad (4, 18), lo cual contrasta con el pronóstico general de la sarcoidosis, ya que cerca de 20% de los pacientes tienen síntomas clínicos permanentes, debido a una fibrosis irreversible, principalmente fibrosis pulmonar, lo que provoca que los pacientes con sarcoidosis tengan una sobrevida más baja que la población general (10, 13, 19).
Aunque en el caso clínico se hizo, no es indispensable la realización de biopsia de piel para establecer el diagnóstico del síndrome de Löfgren (17).
En relación con el tratamiento no hay cura para la sarcoidosis, y el tratamiento sólo cambia el proceso granulomatoso y su consecuencia clínica (10). Existe debate, si el tratamiento puede cambiar el curso natural de la enfermedad, particularmente la fibrosis (6, 10, 14).
La decisión de tratar ya sea inmediatamente o durante un periodo de seguimiento, depende de tres factores: riesgo de severa disfunción o daño irreversible de órganos mayores, riesgo de muerte, o la presencia de síntomas constitucionales incapacitantes (10).
La mejor estrategia puede ser observar al paciente por la alta probabilidad de resolución espontánea, y decidir un tratamiento cuando la enfermedad progrese (10).
Conclusión
Se presenta una paciente con un diagnostico confirmado de síndrome de Löfgren, se resalta el enfoque clínico en pacientes con eritema nodoso y artralgias como principales manifestaciones, y se indica la importancia de tener en cuenta la sarcoidosis en el diagnóstico diferencial etiológico. Además de mostrar la importancia de una adecuada historia clínica, un examen físico completo y una valoración global del paciente lo cual permite llegar a diagnósticos complejos como el caso expuesto.
En pacientes que se presentan con eritema nodoso, la sarcoidosis es una entidad que se debe tener siempre en cuenta al igual que su variante el síndrome de Löfgren a pesar de ser entidades de baja prevalencia en nuestro país.
Referencias
1. Löfgren S, Lundback H. The bilateral hilar lymphoma syndrome; a study of the relation to tuberculosis and sarcoidosis in 212 cases. Acta Med Scand 1952;142: 265-73. [ Links ]
2. Petrilla J. Löfgren syndrome: A clinical variant of sarcoidosis. Hospital Physician2002: 40-3. [ Links ]
3. Bourdillon L, Lanier-Gachon E, Stankovic K. Löfgren syndrome and peritoneal involvement by sarcoidosis. Chest 2007; 132: 310-2. [ Links ]
4. Grunewald J, Eklund A. Löfgren syndrome. Human leukocite antigen strongly influences the disease course. Am J Respir Crit Care Med 2009; 179: 307-12. [ Links ]
5. Wiken M, Ostadkarampour M, Eklund A. Antigen-specific multifunctional T-cells in sarcoidosis patients with Löfgren syndrome. Eur Respir J 2012;40: 110-21. [ Links ]
6. Rao DA, Dellaripa PF. Extrapulmonary manifestations of sarcoidosis. Rheum Dis Clin North Am 2013; 39: 277-97. [ Links ]
7. Schwartz R, Nervi S. Erythema nodosum: A sign os systemic disease. Am Fam Physician 2007; 75: 695-700. [ Links ]
8. Blake T, Manahan M, Rodins K. Erythema nodosum. A review of an uncommon panniculitis. Dermatology Online Journal 2014; 20(4): 3. [ Links ]
9. Mana J, Morcoval J. Erythema nodosum. Clinics in Dermatology 2007; 25: 288-94. [ Links ]
10. Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet P-Y, Müller-Quernheim J. Sarcoidosis. Lancet 2014; 383 (9923): 1155-67. [ Links ]
11. Valovis R. Sarcoidosis estudio clínico de 51 casos y revisión de la literatura. Act Med Colomb 1977; 2:101-10. [ Links ]
12. Costabel. U. Sarcoidosis: clinical update. Eur Respir J 2001;18 (suppl. 32) 56s68s. [ Links ]
13. Haimovic A, Sanchez M, Judson M. Sarcoidosis: a comprehensive review and update for the dermatologist. Part I. Cutaneous disease. J Am Acad Dermatol 2012; 66:5: 699.e1-699.e18. [ Links ]
14. Sehgal VN, Riyaz N, Chatterjee K, Venkatash P, Sharma S. Sarcoidosis as a systemic disease. Clin Dermatol 2014; 32(3):351-63. [ Links ]
15. Morgenthau AS, Iannuzzi MC. Recent advances in sarcoidosis. Chest 2011; 139(1):174-82. [ Links ]
16. Heinle R, Chang C. Diagnostic criteria for sarcoidosis. Autoimmun Rev; 2014; 13(4-5): 383-7. [ Links ]
17. Villanueva G, Kurnat Y, Vasquez J. Síndrome de Löfgren. A propósito de un caso. Semergen 2009; 35(5): 249-51. [ Links ]
18. Kobac S, Yalcin M, Sever F. Sarcoidosis presenting as Löfgren syndrome with myopathy. Case Reports in Rheumatology 2013; Article ID 125251, 3 pages. [ Links ]
19. Haimovic A, Sanchez M, Judson M. Sarcoidosis: a comprehensive review and update for the dermatologist. Part II. Extracutaneous disease. J Am Acad Dermatol 2012; 66(5):719.e1-719.e10. [ Links ]

