










Servicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActa Medica Colombiana
versión impresa ISSN 0120-2448
Acta Med Colomb vol.44 no.3 Bogotá jul./set. 2019
https://doi.org/10.36104/amc.2019.1207
PRESENTACIÓN DE CASOS
Hemofilia adquirida en un anciano
1Geriatra, Hospital Universitario San Ignacio, Programa de Postgrado Pontificia Universidad Javeriana.
2Geriatra, Internista, Epidemiólogo, Hospital Universitario San Ignacio, Programa de Postgrado Pontificia Universidad Javeriana, Bogotá, D.C. (Colombia).
La hemofilia adquirida es un trastorno hemostásico poco frecuente pero altamente fatal que se presenta predominantemente en personas ancianas. Es un trastorno secundario al desarrollo de autoanticuerpos específicos dirigidos contra el factor de coagulación VIII. Se caracterizan por debutar hemorragias potencialmente fatales a nivel gastrointestinal, pulmonar, retroperitoneal, de tejidos blandos o intracraneal, por lo que requiere diagnósticos tempranos y tratamientos eficaces para su tratamiento. El presente caso es de un hombre de 78 años con hemorragia gastrointestinal asociado a equimosis y hematomas de aparición súbita en tejidos blandos y como complicación presenta hematoma laríngeo, con tiempo parcial de protrombina (PTT) prolongado, niveles de factor VIII elevados y niveles de inhibidor de factor VIII elevados. (Acta Med Colomb 2019; 44. DOI:https://doi.org/10.36104/amc.2019.1207).
Palabras clave: hemofilia adquirida; factor VIII; inhibidor; coagulopatía
Acquired hemophilia is a rare but highly fatal hemostatic disorder that occurs predominantly in elderly people. It is a disorder secondary to the development of specific autoantibodies directed against coagulation factor VIII. It is characterized by potentially fatal gastrointestinal, pulmonary, retroperitoneal, soft tissue or intracranial hemorrhages, so it requires early diagnoses and effective treatments. The present case is of a 78-year-old man with sudden onset gastrointestinal hemorrhage associated with ecchymosis and hematomas in soft tissues, with the complication of a laryngeal hematoma. He had a prolonged partial thromboplastin time (PTT), elevated factor VIII levels and elevated factor VIII inhibitorst. (Acta Med Colomb 2019; 44. DOI:https://doi.org/10.36104/amc.2019.1207).
Keywords: acquired hemophilia; factor VIII; inhibitor; coagulopathy
Introducción
La hemofilia adquirida es un trastorno hemorrágico secundario al desarrollo de autoanticuerpos específicos que inhiben a alguno de los factores de la coagulación 1, ocurre espontáneamente en individuos con hemostasia previa normal, no hemofílicos, o sin historia familiar de discrasias sanguíneas 2. El factor VIII (FVIII) es la diana más frecuente de estos autoanticuerpos, denominada hemofilia adquirida A 3, pero también se pueden generar factores inhibidores de otros componentes de la coagulación, que neutralizan la función específica del factor de coagulación al que va dirigido, generando una diátesis hemorrágica 4, que conducen a episodios hemorrágicos graves que se producen hasta en 90% de los pacientes afectados y se asocian a mortalidad que ocurre en 8 - 22% de los pacientes 2,5-8.
Es más prevalente en pacientes ancianos, hombres, con una relación de riesgo directamente proporcional a la edad, así entre los 65 y 85 años se presentan nueve casos por millón de habitantes y en los mayores de 85 años, 15 casos por millón 9-22.
El desconocimiento de la presentación de la enfermedad, del proceso diagnóstico y la severidad de la presentación contribuye a la alta tasa de morbilidad y mortalidad 9. Así mismo, la presentación clínica y su severidad no se correlaciona con el nivel de factor VIII o el título del inhibidor, constituyendo una falla en el pronóstico, y forjando un riesgo de sangrado espontáneo permanente y potencialmente mortal hasta que el inhibidor haya sido erradicado 1,13. Presentamos el caso de un paciente anciano hombre que debuta con manifestaciones hemorrágicas secundarias a la presencia de factor inhibidor de factor VIII, con múltiples morbilidades que modifican el fenotipo clínico de la enfermedad y condicionan su tratamiento.
Reporte de caso
Paciente masculino de 74 años quien consulta por presentar cuadro de 12 horas de episodios de hematoquecia y posteriormente rectorragia en tres oportunidades asociado a dolor abdominal difuso de moderada intensidad.
Como antecedentes patológicos refiere hipertensión arterial en manejo farmacológico con ácido acetil salicílico 100 mg/día, atorvastatina 40 mg/día y enalapril 20 mg/día. Sin otros antecedentes. En la revisión por sistemas comenta aparición de múltiples equimosis en extremidades desde hace 15 días.
Funcionalmente es independiente para las actividades básicas e instrumentales de la vida diaria. No refiere quejas de memoria y se aplicó la escala de mini mental test con un resultado de 30/30, no manifiesta alteraciones en el ánimo, y tiene una adecuada red de apoyo.
En el examen físico de ingreso presentaba estabilidad hemodinámica, conjuntivas hipocrómicas, equimosis orofaríngea. Cuello con equimosis en región anterior cervical. En tórax presenta equimosis en región esternal y supraclavicular bilateral. Ruidos cardiacos taquicárdicos, sin soplos. Ruidos respiratorios normales. Abdomen normal, al tacto rectal restos de melenas. Extremidades con múltiples equimosis en miembros superiores e inferiores. A nivel neurológico sin alteraciones.
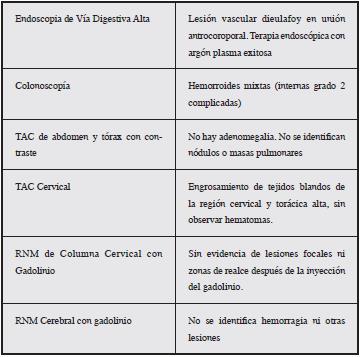
En los paraclínicos se documenta hemoglobina de 6.4 g/dL, con endoscopia de vías digestivas altas que reportó malformación vascular antral (Dieulafoy), tratada con escleroterapia y argón plasma y se indicó terapia con inhibidor de bomba de protones. Colonoscopia con enfermedad hemorroidal, sin sangrados.
Presentó PTT prolongado, sin compromiso de otras líneas hematológicas. C3 y C4 normales, ANAS negativo, coombs directo negativo, anticoagulante lúpico negativo. PTT cruzado con patrón que no corrige pre y post incubación, positivo para anticoagulante circulante, sin estigmas de autoinmunidad. Extendido de sangre periférica con microcitos e hipocromía (Tabla 1).
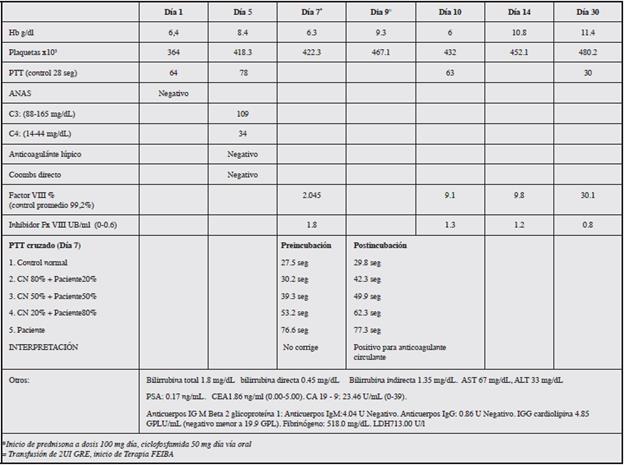
Siete días después del ingreso presenta taquicardia, hipotensión arterial, taquipnea y esfuerzo respiratorio, extensa equimosis en región esternal y cervical y anemización de 2 gr de hemoglobina, sin evidencia de sangrado activo. Fue valorado por hematología, quienes, por evidencia de prueba de mezclas compatible con inhibidor de la coagulación, y anticoagulante lúpico negativo, en ausencia de estigmas de autoinmunidad, debe descartarse la presencia de inhibidor específico (hemofilia adquirida).
Los resultados de estudios sugieren hemofilia adquirida con déficit moderado de factor VIII (2.5%), considerándose inicio de prednisona a dosis 100 mg día, ciclofosfamida 50 mg día vía oral y ampliar estudio en búsqueda de patología primaria como causa de coagulopatía secundaria.
Durante su evolución presenta disfonía, estridor leve, disfagia y desaturación progresiva, crecimiento de equimosis y hematoma en región anterior del cuello y tórax. Los paraclínicos evidencian disminución de 3 gr de hemoglobina a pesar de terapia transfusional Se realizó nasofribrolaringos-copia con evidencia hematoma faríngeo con compromiso de la luz esofágica y de la vía aérea. Se decide inicio de terapia de Bypass con FEIBA, la cual se administró durante 28 días con una dosis promedio ajustada de 4000 UI cada ocho horas (60 UI/KG dosis), con lo que se logró adecuado control hemostático, realizando ajuste de dosis según respuesta (Tabla 2). Se realiza control paraclínico de niveles de factor VIII 30%, sin anemización, además se descartó posible patología neoplásica y autoinmune asociada.
Discusión
Ante un paciente anciano con sospecha de coagulopatía, se debe establecer una secuencia diagnóstica que lleve a la determinación de la causa para instaurar la estrategia terapéutica adecuada. Para comenzar es necesario iniciar con la historia clínica detallada que incluya el uso de medicamentos antitrombóticos, la presencia de enfermedades que alteren las células y las proteínas de la cascada de la coagulación y la ausencia de historia familiar de trastornos hemorrágicos, asociado a la aparición de lesiones hemorrágicas o hemorragias severas de aparición aguda con ausencia de alteraciones a nivel de las células hematológicas, y evidencia de PTT prolongado de manera inexplicable, se debe considerar el diagnóstico de coagulopatía por presencia de factor inhibidor o hemofilia adquirida 12,14.
El paciente expuesto debutó con equimosis y hematomas en tejidos blandos con posteriores hemorragias con alto riesgo de mortalidad, como lo son la hemorragia de vías digestivas y el hematoma laríngeo. La presentación clínica típica de la hemofilia adquirida cursa con hemorragias subcutáneas generalizadas en el 80% de los casos, hemorragias gastrointestinales y mucosas, hematomas musculares, lesión neurovascular y hemorragia del tracto urogenital 1,2,7.
El proceso diagnóstico depende de la experticia del clínico en el enfoque del síndrome coagulopático. Como se identificó en el caso, es necesaria la realización de pruebas de PTT cruzado y cuantificación del factor VIII, con el fin de identificar factores inhibidores y déficit del factor contra el cual van dirigidos los autoanticuerpos 15.
La etiología del desarrollo de autoanticuerpos que inhiben el factor VIIII, es en la mayoría de los casos idiopática, habiendo descartado enfermedades autoinmunes como artritis reumática, lupus eritematoso sistémico, la polimialgia reumática, hipo e hipertiroidismo 1,6,16.
Además, enfermedades linfoproliferativas como leucemia crónica y el mieloma múltiple; los carcinomas tumorales sólidos de pulmón, mama y páncreas 17,18. También, enfermedades dermatológicas como penfigoide psoriasis y pénfigo vulgar; medicamentos como penicilinas, sulfonamidas, fenitoína, interferón alfa y rituximab 19, entre otros; condiciones inflamatorias pulmonares como asma y EPOC 1,2,16,20. En el caso de este paciente se realizó un estudio completo descartándose dichas causas, concluyendo el origen idiopático del cuadro coagulopático.
Una vez hecho el diagnóstico se debe instaurar el tratamiento orientado al control del sangrado, si el paciente lo requiere transfundir hemoderivados, abordar tempranamente las complicaciones, evitar procedimientos invasivos, erradicar el inhibidor y tratar la enfermedad subyacente 1,13,21.
Las terapias hemostáticas utilizadas en la hemofilia adquirida se implementan gracias a su eficacia y la necesidad de lograr rápidamente hemostasia 4,21.
Las guías actuales recomiendan el uso del factor VII recombinante activado (rFVIIa), el concentrado de protrom-bina activado de complejo de derivado de plasma (pd-aPCC) y el factor de anulación de la actividad de inhibidor VIII [FEIBA] como terapia hemostática de primera línea 1,21,22. En el paciente del caso se utilizó esta última, logrando adecuada respuesta tras 21 días de manejo, con seguimiento de niveles de hemoglobina, PT y factor VIII. La terapia erra-dicadora del inhibidor ha comprobado disminuir mortalidad y morbilidad en los pacientes, además disminuye el tiempo de hospitalización y el requerimiento transfusional 2,7,8.
Se pueden usar los corticoides solos o en compañía de ciclofosfamida o rituximab para este objetivo. A pesar de esto la posibilidad de recaída en los pacientes en muy alta, en especial con los pacientes ancianos 9,20.
References
1. Kruse-Jarres R, Kempton CL, Baudo F, Collins PW, Knoebl P, Leissinger CA, et al. Acquired hemophilia A: Updated review of evidence and treatment guidance. Am J Hematol. 2017; 92(7):695-705. [ Links ]
2. Collins P, Baudo F, Huth-Kuhne A, Ingerslev J, Kessler CM, Castellano ME, et al. Consensus recommendations for the diagnosis and treatment of acquired hemophilia A. BMC Res Notes. 2010; 3:161. [ Links ]
3. Franchini M, Gandini G, Di Paolantonio T, Mariani G. Acquired hemophilia A: A concise review. Am J Hematol. 2005; 80(1):55-63. [ Links ]
4. Ma AD, Carrizosa D. Acquired Factor VIII Inhibitors: Pathophysiology and Treatment. Hematology. 2006; 2006(1):432-7. [ Links ]
5. Franchini M, Lippi G. How I treat Acquired factor VIII inhibitors. Blood. 2008; 112(2):250-5. [ Links ]
6. Knoebl P, Marco P, Baudo F, Collins P, Huth-K??hne A, Nemes L, et al. Demographic and clinical data in acquired hemophilia A: Results from the European Acquired Haemophilia Registry (EACH2). J ThrombHaemost. 2012; 10(4):622-31. [ Links ]
7. Collins PW, Hirsch S, Baglin TP, Dolan G, Hanley J, Makris M, Keeling DM, Liesner R, Brown SA, Hay CR, et al. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors' Organisation. Blood.2006; 109(5):1870-7. [ Links ]
8. Baudo F, De Cataldo F. Acquired hemophilia: A critical bleeding syndrome. Haematologica. 2004; 89(1):96-100. [ Links ]
9. Coppola A, Favaloro MDEJ, Ph D, Rcpa FFS, Tufano A, Matteo PD, et al. Acquired Inhibitors of Coagulation Factors: Part I - Acquired Hemophilia A. Semin Thromb Hemost. 2012; 1(212):433-16. [ Links ]
10. Ceresetto JM, Duboscq C, Fondevila C, Pinto MT. Hemofilia adquirida (inhibidor adquirido del factor VIII). Med. 2015; 75(4):231-8. [ Links ]
11. Woods S, Varghese B. Acquired hemophilia A presenting in an elderly man. Cmaj. 2007; 177(4):341-2. [ Links ]
12. Tufano A, Coppola A, Guida A, Cimino E, De Gregorio AM, Cerbone AM, et al. Acquired haemophilia A in the elderly: Case reports. Curr Gerontol Geriatr Res. 2010; 2010(Table 1). [ Links ]
13. Delgado J. Acquired Haemophilia: Review and Meta-Analysis Focused on Therapy and Prognostic Factors. Br J Haematol. 2003; 121:21-35. [ Links ]
14. Hunt BJ. Bleeding and Coagulopathies in Critical Care. N Engl J Med. 2014; 370(9):847-59. [ Links ]
15. Kruse-Jarres R, Quon D, Schramm W, Dolan G. Management of bleeding disorders in the elderly. Haemophilia. 2012; 18(SUPPL. 2):37-45. [ Links ]
16. Janbain M, Leissinger CA, Kruse-Jarres R. Acquired hemophilia A: emerging treatment options. J Blood Med. 2015; 6:143-50. [ Links ]
17. Sallah S, Wan JY. Inhibitors against factor VIII in patients with cancer: Analysis of 41 patients. Cancer. 2001; 91(6):1067-74. [ Links ]
18. Escobar MA. Bleeding in the patient with a malignancy: Is it an acquired factor VIII inhibitor? Cancer. 2012; 118(2):312-20. [ Links ]
19. Franchini M. Rituximab in the treatment of adult acquired hemophilia A: A systematic review. Crit Rev Oncol Hematol. 2007; 63(1):47-52. [ Links ]
20. Rivera Cora NI, Irizarry Delgado F, Merle Ramírez SM, Vera Quiñones J. Acquired Hemophilia A in an advanced age patient of hispanic origin: a case report. BMC Res Notes. 2017; 10(1):432-38. [ Links ]
21. Kessler CM, Knöbl P. Acquired haemophilia: An overview for clinical practice. Eur J Haematol. 2015; 95:36-44. [ Links ]
22. Borg JY, Négrier C, Durieu I, Dolimier E, Masquelier AM, Lévesque H, et al. FEIBA in the treatment of acquired haemophilia A: Results from the prospective multicentre French "FEIBA dans l"hémophilie A acquise' (FEIBHAC) registry. Haemophilia. 2015; 21(3):330-7. [ Links ]
Recibido: 11 de Abril de 2018; Aprobado: 22 de Julio de 2019
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
