











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Wilson's disease, or lenticular degeneration, is a rare genetic disorder that affects copper metabolism, resulting in its abnormal accumulation in the liver, brain, cornea and kidney 1,2. It is globally distributed, with an estimated incidence of 30 cases per million inhabitants, typically presenting between the first and fourth decade of life, although it ranges anywhere from two to 70 years of age 3,4. Although the main symptoms of the disease are hepatic and neurological, Coombs-negative hemolytic anemia may be the only manifestation, and it tends to present months before clinically evident liver disease or neurological manifestations 5. We present the case of a young patient, with no significant medical history, who was admitted to the emergency room with Coombs-negative hemolytic anemia, and was diagnosed with Wilson's disease after several tests.
Clinical case description
A 16-year-old patient was treated for parasites with metronidazole, secnidazole and albendazole three weeks prior to her admission. She consulted due to a two-week history of progressively worsening asthenia, weakness and fatigue, with no other abnormalities or signs. Her guardians stated that after one week she developed progressive pallor and the initial symptoms worsened, adding a throbbing generalized headache with an intensity of 6/10, triggered by exertion, without nausea, vomiting, photophobia or other symptoms. She reported that along with the pallor, she noticed changes in the color of her urine; it was yellow at first and then turned orange. During the two days prior to admission, her symptoms progressed, forcing her to stay home; thus, they decided to take her to our hospital. She denied chest pain, dyspnea, fever, weight loss, skin lesions and other symptoms. This was the first episode of the disease and she had not had other important events which could be related to the current clinical picture.
Her admission lab exams revealed severe macrocytic anemia, thrombocytopenia and hyperbilirubinemia with increased direct and indirect fractions. A peripheral blood smear showed macrocytes and polychromatophilia, with no other relevant findings. Hemolysis studies revealed elevated LDH, decreased haptoglobin and a negative fractionated Coombs. Aspartate transaminase (AST) was also elevated with normal alanine transaminase (ALT), decreased alkaline phosphatase, prolonged INR and coagulation times, and hypoalbuminemia. Kidney function tests and electrolyte levels were within normal limits and the infectious disease panel was negative (Table 1). Abdominal ultrasound and abdominal magnetic resonance showed a normal-sized liver with a nodular surface, suggesting hepatic cirrhosis. During her hospitalization, the laboratory findings persisted and towards day three, she developed a decreased attention span and sleep disturbances, without asterixis, which, together with an INR >1.5 established the diagnosis of acute liver failure in a patient with cirrhosis.
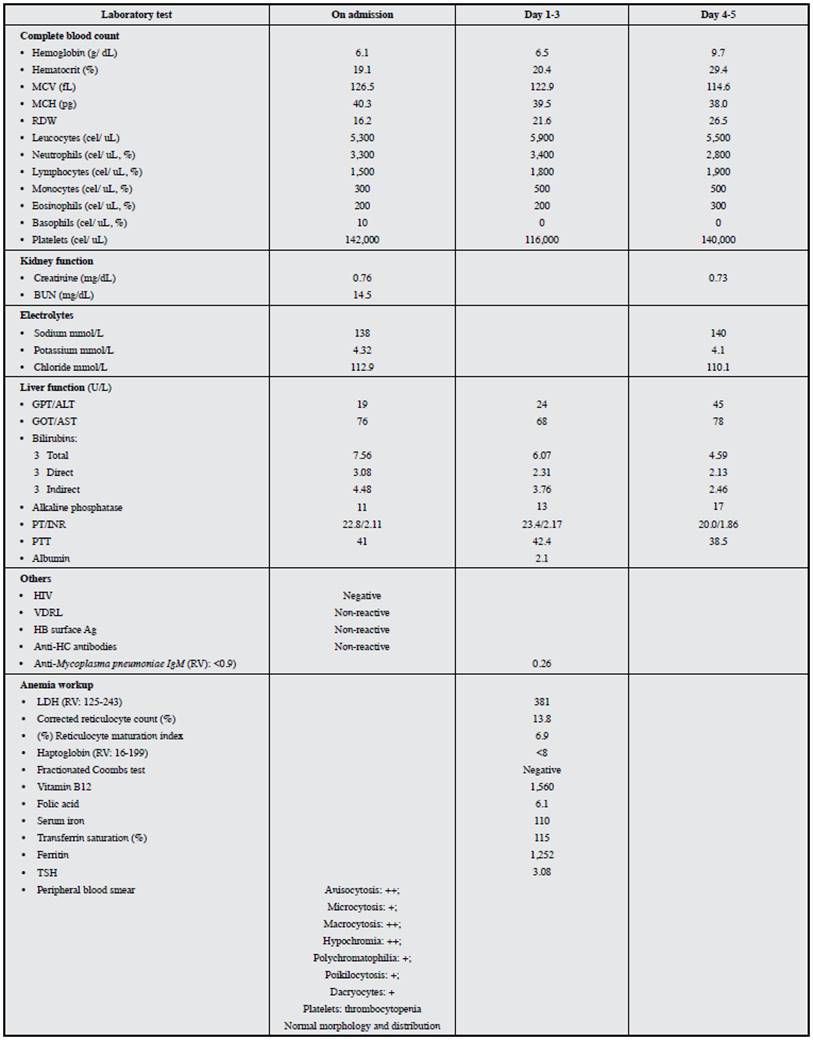
In view of non-immune hemolytic anemia in a young woman with cirrhosis of unknown etiology, and considering the alkaline phosphatase/total bilirubin ratio <4 and AST/ALT >2.2, diagnostic tests were run for suspected Wilson's disease, which showed decreased ceruloplasmin levels and increased 24-hour urine copper. A slit lamp test showed Kayser-Fleischer rings (Table 2), which established the diagnosis of acute liver failure secondary to Wilson's disease.

Discussion
This case presents an adolescent woman with no prior medical history or illness. Wilson's disease is a rare disorder which typically presents between the first and fourth decade of life and does not generally manifest clinically before five years of age, since it takes time for the copper to accumulate 6. The disorder is caused by autosomal recessive mutations of the ATP7B gene on chromosome 13. This gene codes for a transmembrane protein in the hepatocyte which ensures copper transport in the trans-Golgi compartment for incorporation into apoceruloplasmin, subsequent formation of ceruloplasmin and elimination of excess copper through the biliary system 7,8. A defect in this gene leads to copper accumulation in the liver and extrahepatic tissues 7
Wilson's disease has a broad clinical presentation and awareness of its manifestations is especially important 9, recognizing that the most frequent symptoms are mainly hepatic and neuropsychiatric 10. Generally speaking, the clinical presentation of this case corresponds to an acute presentation of the disease, which is characterized by liver involvement, hemolysis, or both, while the chronic form involves chronic liver disease, neuropsychiatric manifestations, or a combination of these 9. In 40-70% of patients, the liver is involved first, and manifestations may include asymptomatic transaminase elevation, chronic liver disease, cirrhosis or acute liver failure 10. On the other hand, neurological and neuropsychiatric disorders may be the presenting symptoms in 40-50% of patients, while hemolytic anemia is uncommon and has been described as the initial symptom in <10-15% of those affected 4,6,9.
Hemolytic anemia, as in this case, tends to be Coombs negative; it frequently remits and may occasionally recur 9,10. During the inpatient course, acute liver failure was diagnosed along with the hemolytic anemia, which has been recognized as the characteristic trait of acute liver failure due to Wilson's disease and may be the first clinical manifestation of the disease in approximately 5% of patients 9,10. Although the exact mechanism of the hemolysis is unknown, it is presumed to be due to the direct toxic effect of excess copper on the surface of the erythrocytes, damaging the cell membrane, oxidizing the hemoglobin and inactivating the pentose phosphate enzyme pathways along with others involved in glycolysis 3,5-8,10.
In this patient, and as recognized in the literature, deep jaundice, low hemoglobin, mild transaminase elevation and low serum alkaline phosphatase strongly suggested the diagnosis of fulminant Wilson's disease 11. In this regard, an alkaline phosphatase/total bilirubin ratio <4 and AST/ALT >2.2 have a 94% sensitivity and 86% specificity, with a likelihood ratio (LR) of 7, for the diagnosis of acute liver failure due to Wilson's disease, which supported this diagnosis in our patient. In this clinical scenario, urinary copper excretion is markedly elevated, while ceruloplasmin and serum copper are less sensitive and specific 3,11.
The diagnosis of this disease is based on a combination of clinical and laboratory findings. Kayser Fleischer rings are present in 50-60% of patients with isolated liver disease, and up to 90% of patients with neurological involvement, explained by copper deposits in Descemet's membrane in the cornea. The slit-lamp test is diagnostic, and although these rings are almost always considered to be pathognomic of the disease, they may also be seen in other chronic cholestatic diseases 3,4,7,11. Ceruloplasmin levels are decreased in >85% of patients, due to defective copper inclusion in this protein which causes it to be eliminated more rapidly. However, a low ceruloplasmin level does not in itself establish a diagnosis of Wilson's disease, since it may also be altered in malnutrition and other diseases 3,7. The release of copper from the hepatocytes increases its 24-hour urinary excretion, which in some patients may reach up to 600-1,600 nmol/day 3,7,11.
A ceruloplasmin level <20 mg/dL associated with Kayser-Fleischer rings or a urine copper level >100 micrograms/day is considered to be diagnostic for the disease. However, 24-hour urinary copper excretion may be normal in close to 25% of patients 3,4,11. In the presented case, the diagnosis was established by a combination of clinical, laboratory and imaging findings, such as Kayser-Fleischer rings, acute liver failure, cirrhosis of unknown etiology, and non-immune hemolytic anemia associated with ceruloplasmin levels <20 mg/dL and elevated urine copper excretion. In conclusion, it may be suggested that in young patients who present with acute liver disease associated with Coombs-negative hemolysis, the possibility of Wilson's disease should be proposed and tests performed to allow an early diagnosis.

