











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Hypertrophic cardiomyopathy is the most common cause of hereditary cardiomyopathy, affecting one in 500 people 1. Hypertrophic cardiomyopathy may involve the whole heart or just the basal, mid-ventricular or the apical segments of both ventricles. The apical type is rare, usually involving the left ventricle and rarely the right ventricle or both 2. It was initially described in Japan in 1976, and is characterized by an "ace of spades" configuration of the left ventricle at end-diastole on ventriculography 3. Historically, it was thought that this condition was restricted to the Japanese population, where it is most prevalent. Out of all the patients with hypertrophic cardiomyopathy in Japan, the incidence of the apical variant was 15%, while in the United States it is 3% 2.
It has a heterogenous clinical presentation and the electro-cardiographic finding of a giant T-wave inversion has been reported as the typical manifestation of this disease. Its diagnosis by imaging using echocardiography may be limited in apical assessment. Therefore, cardiac magnetic resonance (MR) has become the non-invasive alternative of choice, replacing ventriculography which was considered the diagnostic gold standard 4. The objective of this case report is to describe a rare variant of a genetic disease with potential cardiovascular morbidity and mortality.
Case report
A 59-year-old male patient with no significant medical history or family history of sudden death or evident cardio-vascular disease was incidentally examined clinically and electrocardiographically (he consulted after accidentally falling from a chair, sustaining a soft tissue chest injury). He occasionally had short episodes of palpitations without syncope, was in NYHA functional class I, and had no heart disease symptoms.
A resting electrocardiogram revealed a ventricular repolarization disorder with deep T-wave inversion on V2 and V3, a normal corrected QT and left ventricular hypertrophy (Figures 1 and 2).
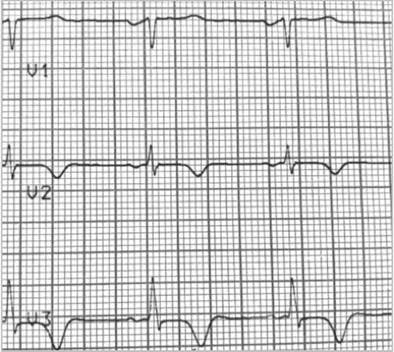
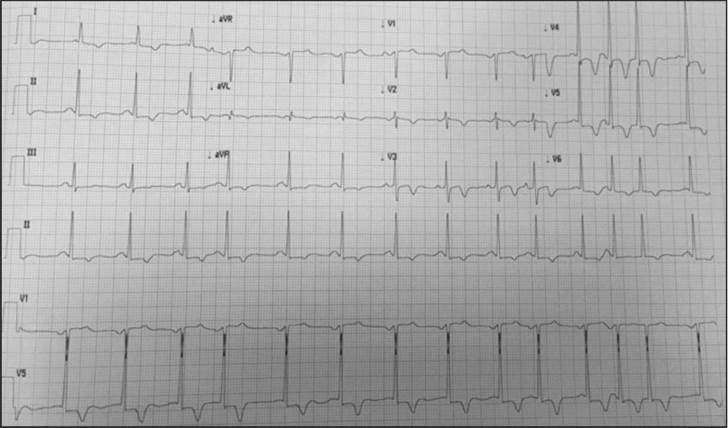
Figure 2 Signs of left ventricular hypertrophy with altered repolarization and inverse T in the precordial leads. Supraventricular extrasystole and doublet.
A 24-hour Holter test was performed due to palpitations, with evidence of frequent premature atrial complexes and nonsustained atrial tachycardia.
The findings of the resting echocardiogram indicated left ventricular (LV) apical hypertrophy, which pointed toward the baseline electrocardiographic findings (apical hypertrophic cardiomyopathy). The noninvasive coronary ischemia study (exercise stress echocardiogram) was negative for inducible ischemia during exercise and the patient did not have increased gradients in the LV outflow tract.
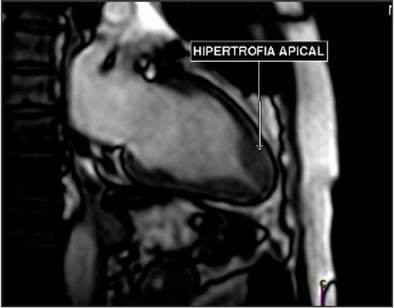
A cardiac MR (Figure 3) confirmed asymmetric apical hypertrophy, with a diastolic apical wall thickness of 17 mm and an apical wall/posterior wall thickness ratio of 1.54, with preserved segmental dynamics and global systolic function. No obstructive gradients were found in the left ventricle.
Hypertrophic cardiomyopathy studies were performed, ruling out arterial hypertension, and screening for Fabry disease was negative. Symptomatic treatment with beta blockers was proposed for his palpitations and frequent atrial extrasystoles.
Given the lack of family history of sudden cardiac death and the low (2.5%) risk of sudden death calculated using the SCD HM scale, he was not considered to be a candidate for an implantable automatic defibrillator. The patient is currently asymptomatic, with a negative study for HCM in first-degree relatives.
Discussion
This case illustrates the silent character of certain diseases which may have significant morbidity and mortality if early intervention is not prescribed. Apical hypertrophic cardio-myopathy as a variant of the sarcomere mutation, known as Yamaguchi syndrome, is a rare morphological variant in which there is no left ventricular outflow tract obstruction, but rather midventricular obstruction 5. In our case, the disease finding was incidental and was diagnosed at the age of 59. Most patients present with symptoms in their fourth decade of life, with a mean age at presentation around 41.4 ± 15 years, and it is more common in males2. Our patient was diagnosed at an older age than that reported in the literature since he was asymptomatic, and although he occasionally experienced palpitations, they were mild and were never studied.
Overall, more than half the patients are asymptomatic, as was the case with our patient's diagnosis. However, those who have symptoms may debut with chest pain, palpitations, heart failure symptoms, dyspnea, atrial fibrillation or syncope 5.
Clinical suspicion begins with a baseline electrocardiogram. The typical electrocardiographic characteristics are ventricular hypertrophy, giant inverse T-waves (>10 mm), ST depression and inverse U waves on DII, III and aVF, V4-V6, and prolonged QTc. In the current case, these findings are compatible with the diagnosis. The most common findings are the classic deep T-wave inversion in the precordial leads in up to 93% of patients and close to 65% of these have left ventricular hypertrophy. Giant inverse T-waves have been reported in 47% of patients with this condition 5. However, it has been reported that giant inverse T-waves are not always seen in this entity, as these, as well as ST segment depression may vary over time 6.
The diagnosis depends on detecting localized apical hypertrophy through imaging, and echocardiography has been the first-line choice for this. However, it has limitations in correctly viewing the apex and its hypertrophy, and therefore this can be missed. These limitations are overcome with a cardiac MR, even in cases where they were missed on echocardiography 7. The diagnostic criteria on resonance imaging are: apical wall thickness greater than 15 mm and/or a left ventricular apical: basal wall thickness ratio > 1.3-1.5 8.
Hypertrophic cardiomyopathy is a diagnosis of exclusion, and therefore secondary causes of left ventricular hypertrophy, such as arterial hypertension, valvular or subvalvular aortic stenosis and infiltrative cardiomyopathy must be ruled out. Screening was done for Fabry disease, which has an estimated prevalence of 0.5-1% in hypertrophic cardiomyopathy, and although symmetric and concentric left ventricular hypertrophy is the most typical pattern in this disease, other variants such as apical hypertrophy have also been reported 9. Other patterns of diseases such as amyloidosis or hemochromatosis were not suggested by imaging or MR, and therefore were not investigated.
With regard to treatment, in hypertrophic cardiomyopathy, regardless of symptoms, an optimized lifestyle is recommended, especially related to education on low-intensity aerobic activities 10.
Excessive alcohol consumption, the use of psychoactive substances, dehydration and extreme temperatures should be avoided, as they may trigger syncope or arrhythmias. Initial treatment consists of beta blockers to decrease the heart rate and reduce the obstructive gradient in the left ventricular outflow tract. Myectomy or alcohol septal ablation have been used to alleviate left ventricular outflow tract obstruction, but with limited evidence in the apical variant 5.
Sudden cardiac death is less likely to occur in patients with isolated apical hypertrophic cardiomyopathy, and overall cardiovascular morbidity is lower compared with other hypertrophic cardiomyopathy phenotypes. However, around 25% of people may develop significant late-onset morbidity including heart failure, chest pain, apical fibrosis, the formation of apical aneurysms, cerebrovascular accidents, atrial fibrillation and ventricular tachycardia 11. In Japan, apical HCM appears to be more benign than in Western countries, which tend to have complications like atrial fibrillation and acute myocardial infarction during long-term follow up 7.
The clinical practice guideline and clinical trials support the use of implantable cardioverter-defibrillators (ICDs) for primary prevention of sudden death (SD) in the following cases: 1) a family history of sudden cardiac death, 2) unexplained syncope, 3) asymptomatic non-sustained ventricular tachycardia, 4) abnormal blood pressure response to exercise, and 5) left ventricular wall thickness greater than 30 mm. However, other potential risk factors have been identified, such as left ventricular outflow tract obstruction, extensive late gadolinium enhancement (myocardial fibrosis), apical aneurysm, genetic/multiple mutations, and left ventricular wall thickness between 25-30 mm 12.
Risk stratification can be done using the HCM risk-SCD calculator 13 which incorporates age, the extent of left ventricular hypertrophy, the size of the left atrium, the left ventricular outflow tract gradient, family history of sudden cardiac death, non-sustained ventricular tachycardia, and unexplained syncope to predict the five-year risk of sudden cardiac death 1. This calculator helps determine the need for defibrillator implantation for primary prevention in patients with no red flags. The following decisions are then recommended according to the risk score 12:
For scores greater than 6%, defibrillator implantation should be considered (NE a COR).
Score from 4-6% (may be considered).
Score less than 4%, not usually indicated.
Finally, after being assessed, our patient had a score of 2.5%, not requiring ICD implantation at this time. It should be noted that there are no trials or predictive models to date to guide this aspect specifically in apical HCM. Although the previous scale is based on HCM with different morphological subtypes, it has been thought that it may underestimate the risk in this specific entity, and therefore other factors should be considered 14.
Family screening was negative, which concurs with what has been described in the most recent literature in which few patients reported a positive family history compared with classic HCM, which suggests potential differences in screening and the presence of other etiological factors 14.
Although access to both family and etiological screening for hypertrophic cardiomyopathy tends to be limited in Colombia, in our case, the healthcare entity's process was prompt. However, this highlights an opportunity for improving our country's public health policies in this field to foster diagnosis and timely treatment and to also include a study of this entity to better understand its behavior locally, as even in the current literature it is considered to be the least understood variant 14.
Conclusions
Yamaguchi syndrome is a rare variant of hypertrophic cardiomyopathy in Western countries. It poses a diagnostic, etiologic, prognostic and treatment challenge compared with the other better known, studied and understood HCM variants. Therefore, more research is needed in this field, especially in Western countries where its complications tend to be greater. Primary prevention of sudden death should be considered, just as in the other hypertrophic cardiomyopathies, searching for red flags and calculating risk, along with symptomatic relief of LV obstruction.

