











 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroduction
Histiocytic necrotizing lymphadenitis or "Kikuchi-Fujimoto disease" (KFD) was first described in 1972 on the Asian continent where it is most prevalent. It often presents in people under the age of 40, affects more women than men with a 2:1 ratio, is benign and self-limited, and is diagnosed histopathologically. The treatment of choice in symptomatic patients is nonsteroidal anti-inflammatory drugs (NSAIDs), although at times the disease can be so severe that glucocorticoids are indicated 1,2.
Case presentation
A 20-year-old single man from Barranquilla (Atlántico), residing in Bucaramanga (Santander), who worked at a chicken retailer and had no significant medical history had been sick for nine days with three non-bloody diarrheal stools, with mucus, and non-quantified fever coupled with chills, general malaise, diffuse abdominal pain, abdominal erythema and an approximately 6 kg involuntary weight loss over an unspecified time period. He had been seen multiple times in urgent care, where symptom management had been prescribed. He consulted once again at another institution due to persistent symptoms, and a complete blood count was ordered showing leukopenia and thrombocytopenia. A stool exam, including ova and parasites, was normal, and his chest x-ray was also normal. He was diagnosed with febrile thrombocytopenia and antibiotic treatment was begun with piperacillin/tazobactam and vancomycin. Blood cultures were negative, as was an autoimmune panel: AST and ALT were elevated; IgM for dengue virus drawn on day nine of the disease was negative, and HBsAg, anti-HBc and anti-HCV were non-reactive. Three days after beginning vancomycin he developed a red, macular itchy rash and was referred to our institution for a hematology evaluation.
The physical exam on admission to our institution showed a patient in good general condition, with facial reddening and erythematous macules covering > 90% of the total body surface, including the palms, left anterior cervical and middle anterior submandibular (< 1 cm) adenopathy, 2x1 cm left inguinal adenopathy and right axillary adenopathy. His cardiopulmonary and neurological exams were normal. A right axillary lymph node (RALN) biopsy was ordered due to a suspected lymphoproliferative syndrome; however, necrotizing lymphadenitis was found with no evidence of an infectious or neoplastic inflammatory process (Figure 1). A skin biopsy was taken for possible skin lymphoma, along with a bone marrow biopsy (Table 1), and a consult with infectious disease was requested due to a suspected infectious process. He was also thought to have red man syndrome; vancomycin was discontinued, and four days later the rash had partially disappeared.
An infectious disease consult was performed, and he was considered to have had exposure to Gram negative bacteria and enterobacteria due to his work history. When his fever did not improve by day seven, and thinking of an enterobacteria infection, piperacillin/tazobactam was discontinued, and ceftriaxone was begun; a possible rickettsial infection was also considered, and so doxycycline was begun. Complementary tests were ordered, which did not suggest an active infectious process (Table 1). After the first day of ceftriaxone plus doxycycline, he only had one fever spike, but doxycycline was discontinued due to gastrointestinal intolerance. On the following day he had a fever spike; blood cultures were drawn, ceftriaxone was changed to meropenem to cover possible bacteremia, and a transesophageal echocardiogram was taken to rule out endocarditis, which was normal. Subsequently, the hematology service ordered a CT of the chest, abdomen and soft tissues of the neck, showing bilateral anterior cervical, axillary and inguinal adenopathy. Two days after beginning treatment with meropenem he had no more fever, and the blood cultures were negative on the third day of incubation, so the treatment was discontinued.
The patient had no further febrile episodes for seven days, his pancytopenia improved, and he was hemodynamically stable, so he was discharged on the 18th day of his hospital stay with NSAID treatment for symptoms. On follow up one month later, the patient reported no fever or accompanying symptoms.
Discussion
Kikuchi-Fujimoto disease frequently occurs when there is an underlying condition like systemic lupus erythematosus, which is why its debut without an underlying disease, presenting in a male, and especially in Colombia, is rare 2-4. Our patient's symptoms ranged from the most common, like lymph node enlargement and fever, to the least common, like diarrhea and weight loss 4,5. The cervical lymphadenopathies are usually posterior, with the anterior presentation occurring in 3.3%, as was the case in our patient 6. The tests show evidence ranging from cytopenia and atypical lymphocytes to elevated ferritin, AST, ALT and lactate dehydrogenase 4,6,7. His work history predisposed him to Gram negative bacteria (salmonella and leptospira) and other enterobacteria which may trigger KFD 2,8,9. However, an infectious process was not documented in this patient, and he received effective antibiotic treatment for these bacteria anyway. His blood cultures were negative, leptospira IgM was negative, and the rest of the tests showed no evidence of bacterial infection. Serology showed prior exposure to toxoplasma, CMV, and EBV (positive IgG with negative IgM); hepatitis and retrovirus serologies were negative; and viral loads for CMV, EBV and HIV were undetectable, ruling out acute viral infection. An autoimmune or neoplastic component triggering this disease was also ruled out with the tests performed; however, this disease may have an uncertain etiology 10.
The differential diagnosis could include Still's syndrome, although he only met the major criterion of fever and elevated ferritin, according to Fautrel et al.'s 11 criteria. For Yamaguchi et al.'s 12 criteria, he had a fever, along with the minor criteria of negative rheumatoid factor and ANAs, abnormal liver tests and adenopathies. Even so, he did not meet the necessary criteria to confirm this disease. For hemophagocytic syndrome, the patient only met three criteria (fever greater than 38.5°, cytopenias and ferritin > 500 ng/mL) of the five needed for diagnosis 13. Tuberculous lymphadenitis was ruled out as the lymph node biopsy did not show infiltrate suggestive of chronic granulomatous disease, and the Ziehl-Neelsen stain showed no acid-alcohol-fast bacilli; also, the patient showed no lung involvement on the chest images, and bone marrow cultures for mycobacteria were negative after six weeks of incubation 14.
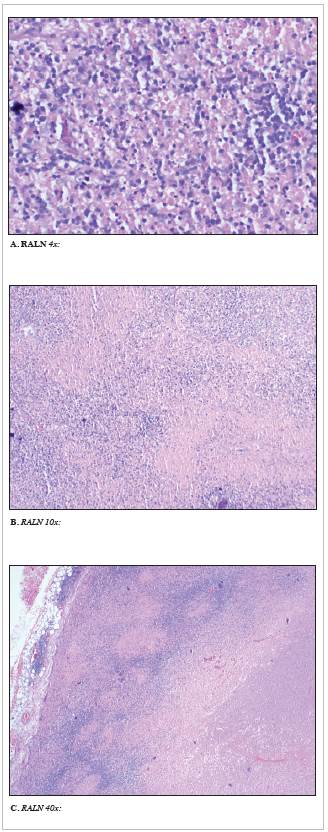
Figure 1 RALN excisional biopsy. Figure 1A, 1B, 1C. Hematoxylin-eosin stain showing necrotizing lymphadenitis changes characterized by geographic areas of fibrinoid necrosis with cellular detritus and frequent apoptotic activity. Special histochemical staining (Ziehl-Neelsen and Gomori) was negative for microorganisms, and immunohistochemical studies were negative for a neoplastic process.
Table 1 Complementary tests.
| Date | Test | Result |
|---|---|---|
| 11/29 | Complete blood count | Hemoglobin: 13.8 gr/dL; leukocytes 1,590/mm3; lymphocytes 430/mm3; neutrophils 1,020/mm3; platelets: 200,000/ mm3 |
| LDH | 660 IU/L | |
| AST | 96 IU/L | |
| ALT | 74 IU/L | |
| Lactic acid | 2.32 mmol/L | |
| Blood cultures x 2 | Negative for microorganism growth at five days. | |
| Peripheral blood smear | Normocytic, normochromic; normal number of leukocytes, normal number of platelets, white and red cell morphology normal. | |
| HBsAg | Non-reactive. | |
| Anti-HCV | Non-reactive. | |
| Total Anti-HBc | Negative. | |
| PT | 36.3 seconds (Daily control: 26.8 seconds) | |
| Urinalysis and urine culture | Urinalysis: density: 1.024, no proteinuria, glycosuria, or hematuria, and negative urine cultures. | |
| 11/30 | VDRL | Non-reactive. |
| 12/1 | HIV | Negative. |
| Triglycerides | 124 mg/dL | |
| Ferritin | 2,552 ng/mL | |
| CMV IgM | Non-reactive. | |
| CMV IgG | Reactive. | |
| Toxoplasma Gondii IgG | Reactive. | |
| EBNA IgM | Non-reactive. | |
| EBNA IgG | Reactive. | |
| 12/2 | CMV viral load | Non-detectable. |
| Leptospira IgM | Non-reactive, taken on day 17 of the disease. | |
| EBV DNA viral load | Non-detectable. | |
| HIV viral load | Non-detectable. | |
| Microstrout Chagas test | Negative. | |
| PT | 37.8 seconds (daily control: 26.8 seconds) | |
| Complete blood count | Hemoglobin: 12.8 gr/dL; leukocytes 1,280/mm3; lymphocytes 217.6/mm3; neutrophils 1,011 uL; platelets: 199,000/mm3 | |
| 12/4 | Complete blood count | Hemoglobin: 13 gr/dL; Türk cell reactive lymphocytes 1%; reactive lymphocytes 18.9 uL; lymphocytes 453.6 uL; neutrophils 1,341.9 uL; platelets: 207,000/mm3 |
| ANAs | Non-reactive. | |
| ESR | 60 mm/1hr | |
| CR/PTT (corrected) | Compatible with factor deficiencies. | |
| 5/12 | Complete blood count | Hemoglobin: 13.5 gr/dL; leukocytes 2,060/mm3; lymphocytes 597.4 uL; neutrophils 1,318.4 uL; plateletes: 217,000/mm3. |
| PT | 32.1 seconds (Daily control: 26.8 seconds) | |
| Bone marrow culture | Tuberculous mycobacteria not detectable in the sample after six weeks' incubation. | |
| Bone marrow aspirate cytometry | Limited cell viability, with no immunophenotypic abnormalities, and 4.44% polyclonal B lymphocytes with normal characteristics. | |
| Bone marrow karyotype | Male chromosomal karyotype with no abnormality in the number and structure of the chromosomes. The conventional cytogenetic study showed a 46 XY chromosomal constitution, with no evidence of structural abnormalities. | |
| Bone marrow biopsy | Hypocellular, with mild/moderate degeneration of the medullary interstitium and no significant morphological alterations of the three hematopoietic series. Mycobacteria undetectable with polymerase chain reaction. Negative culture at 5, 28 and 42 days of incubation. | |
| Irregular antibodies | Negative. | |
| RUE blood cultures x 2 | Negative for any microorganism growth after five days. | |
| 12/11 | Complete blood count | Hemoglobin: 12.6 gr/dL; leukocytes 2,540/mm3; lymphocytes 640/mm3; neutrophils 1,900 uL; platelets: 154,000/mm3. |
| 12/14 | LDH | 278 IU/L. |
| *Abbreviations: RUE: right upper extremity; LDH: lactate dehydrogenase; AST: aspartate aminotransferase; HBsAg: hepatitis B surface antigen; Anti-HCV: anti-hepatitis C virus antibodies; Total anti-HBc: total anti-hepatitis B core antibodies; CMV: cytomegalovirus; EBNA: Epstein Barr nuclear antigen; EBV: Epstein Barr virus; PT: prothrombin time; ANAs: antinuclear antibodies; ESR: erythrocyte sedimentation rate; CR/PTT: crossed partial thromboplastin time. |
Kikuchi-Fujimoto disease is often diagnosed by histopathological lymph node studies showing necrotic foci and fibrinoid necrosis, findings compatible with this patient's biopsy 2,7,15. Lastly, the patient's prognosis is good, and therefore the treatment will be symptomatic. Symptoms may abate in one to four months, recurrence has only been found in 3-4% of cases, and mortality is low (2.1%) 2,15.
In view of the above, and having symptoms compatible with other more prevalent diseases in our region, it is an infrequently suspected disease. Therefore, treatment may be inadequate, and knowledge of this disease is essential, keeping it within the differential diagnosis of patients with characteristics similar to those presented by our patient, with no explainable cause.

