










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkActa Medica Colombiana
versão impressa ISSN 0120-2448
Acta Med Colomb vol.47 no.4 Bogotá jul./dez. 2022 Epub 28-Maio-2023
https://doi.org/10.36104/amc.2022.2558
Presentación de casos
Glomeruloesclerosis focal y segmentaria en paciente con deficiencia congénita de lecitin colesterol aciltransferasa
a Especia lista en Medicina Interna y Nefrología Depar tamento de Nefrología, Clínica Los Rosales;
b Residente de Medicina Interna Universidad Tecnológica de Pereira. Pereira (Colombia).
c Escuela de Medicina Universidad de Nariño. Pasto (Colombia).
La deficiencia de lecitin-colesterol aciltransferasa es una enfermedad genética rara, causada por una mutación en el gen que codifica la proteína lecitin-colesterol aciltransferasa y afecta principalmente el metabolismo de las lipoproteínas de baja densidad. Se manifiesta típicamente con opacidades corneales difusas, anemia normocítica y enfermedad renal. Se presenta el caso de un hombre de 30 años con enfermedad renal crónica y síndrome nefrótico, con biopsia renal inicial que demostró un patrón de glomeruloesclerosis focal y segmentaria, interpretada como primaria, enfermedad que fue refractaria a múltiples esquemas de inmunosupresión. Las manifestaciones como anemia, esplenomegalia, opacidades corneales y la asociación con lipoproteínas de alta densidad bajas, alertaron sobre la posibilidad de compromiso glomerular secundario a un déficit de la enzima lecitin-colesterol aciltransferasa, confirmado mediante estudio de secuenciación genética. Dada la baja incidencia de la enfermedad, el diagnóstico representa un desafío clínico. Las manifestaciones suelen interpretarse como eventos aislados, lo que lleva a retraso significativo en su confirmación. El conocimiento de esta entidad y el ejercicio clínico necesario para llegar al diagnóstico, servirán como referencia que derive en la sospecha y reporte de futuros casos. (Acta Med Colomb 2022; 47. DOI:https://doi.org/10.36104/amc.2022.2558).
Palabras clave: deficiencia de lecitin-colesterol aciltransferasa; glomeruloesclerosis focal y segmentaria; enfermedad renal crónica; anemia; opacidades corneales
Lecithin-cholesterol acyltransferase deficiency is a rare genetic disease caused by a mutation of the gene coding for the lecithin-cholesterol acyltransferase protein, and mainly affects low density lipoprotein metabolism. It typically manifests with diffuse corneal opacities, normocytic anemia and kidney disease. We present the case of a 30-year-old man with chronic kidney disease and nephrotic syndrome. His initial kidney biopsy showed focal segmental glomerulosclerosis, thought to be primary, a disease which was refractory to multiple immunosuppressive schemes. Manifestations such as anemia, splenomegaly, corneal opacities and an association with low high-density lipoproteins alerted to the possibility of glomerular damage secondary to lecithin-cholesterol acyltransferase enzyme deficiency, which was confirmed through genetic sequenc ing. Due to the low incidence of the disease, diagnosis is a clinical challenge. The signs and symptoms tend to be interpreted as isolated events, which significantly delays its confirmation. Understanding this entity and the clinical exercise needed to arrive at its diagnosis will serve as a reference, resulting in the suspicion and reporting of cases in the future. (Acta Med Colomb 2022; 47. DOI:https://doi.org/10.36104/amc.2022.2558).
Keywords: lecithin-cholesterol acyltransferase deficiency; focal segmental glomerulosclerosis; chronic kidney disease; anemia; corneal opacities
Introducción
La deficiencia de lecitin-colesterol aciltransferasa (LCAT) es una enfermedad genética autosómica recesiva poco frecuente, se estima una prevalencia a nivel mundial de 1/1 000 000 habitantes y se presenta con la triada clásica de opacidades corneales difusas, anemia hemolítica y en fermedad renal, esta última representa la principal causa de morbimortalidad, inicia en la infancia, cursa con proteinuria y progresión a enfermedad renal crónica hacia la cuarta o quinta década de la vida y a la fecha no hay tratamiento efectivo disponible, además los pacientes que acceden a un trasplante renal se ha documentado rápida recurrencia de la enfermedad en el injerto 1,2. Hasta el momento en Colombia y Latinoamérica la información reportada sobre esta enfermedad es escasa, por lo que el presente caso aporta al conocimiento sobre la presentación clínica de una enfermedad rara, con un diagnóstico que requiere de un alto índice de sospecha integrando una amplia constelación de signos, síntomas y hallazgos de laboratorio que generalmente son abordados de forma aislada y atribuidos a otras causas.
Reporte de caso
Paciente masculino hispano de 30 años, quien seis años antes consultó por edema en miembros inferiores, los exáme nes revelaron una proteinuria en orina de 24 horas en 4.3 gr, albúmina 2.8 gr/dL, triglicéridos 1388 mg/dL, colesterol total 263 mg/dL, colesterol de alta densidad (HDL) 5.0 mg/dL, colesterol de baja densidad (LDL) 19 mg/dL, creatinina 1.2 mg/dL, uroanálisis con proteinuria 500 mg/dL, sin hematuria, los estudios de infección activa, enfermedad inmunológica y neoplásica fueron negativos. Una biopsia renal reportó glomeruloesclerosis focal y segmentaria (GSFS) interpretada como primaria. Recibió tratamiento inicial con dosis altas de prednisona y mofetil micofenolato, pero ante no mejoría de la proteinuria recibe ciclofosfamida intravenosa, posteriormente ciclosporina por tres años y finalmente tacrolimus sin lograr respuesta.
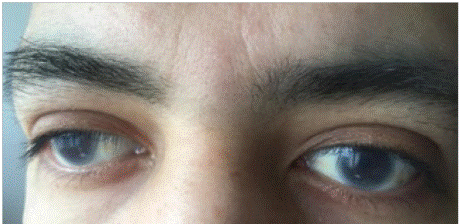
Consultó al servicio de urgencias por dolor en hipocon drio derecho, asociado a náuseas, cinco episodios eméticos y edema persistente en extremidades; sin otros hallazgos relevantes en la revisión por sistemas. Como antecedentes los padres del paciente tienen un vínculo de consanguinidad (primos hermanos). Tiene una hermana de 22 años con dislipidemia y opacidades corneales similares a las del paciente. Los signos vitales fueron normales, en el examen físico se evidenció presencia de opacidades corneales bilaterales, sin hallazgos cardiopulmonares anormales; dolor con la palpación del hipocondrio derecho, sin signos de irritación peritoneal, sin hepatomegalia ni esplenomegalia palpable, edema grado II en miembros inferiores. El resto de la va loración fue normal.
Las pruebas de laboratorio reportaron los siguientes valores: creatinina 2.35 mg/dL, nitrógeno ureico 50 mg/dL, proteinuria en orina de 24 horas 10.2 gr, albumina 1.19 gr/dL, triglicéridos 846 mg/dL, colesterol total 194 mg/dL, colesterol LDL 10 mg/dL, colesterol HDL 13.9 mg/dL, hemog lobina 7.6 gr/dL, hematocrito 20.4%, volumen corpuscular medio 86 fl, el resto del hemograma fue normal, recuento de reticulocitos corregido 0.1%, coombs directo negativo, hierro total 65.9 u/dL, transferrina 125 mg/dL, índice de saturación de transferrina de 36%. La electroforesis de proteínas en suero y la biopsia de medula ósea descartaron paraproteinemia. Los estudios para hepatitis virales, sífilis y virus de inmunodeficiencia humana fueron negativos, al igual que los anticuerpos antinucleares. El complemento C4 fue normal y el C3 se encontró consumido (54.9 mg/dL). La medición de la actividad de alfa galactosidasa descartó enfermedad de Fabry. Una ecografía abdominal demostró presencia de hidrocolecisto y esplenomegalia de 15.9 cm, el tamaño de ambos ríñones fue normal.
El hallazgo de opacidades corneales, anemia, dislipidemia con HDL muy bajo, falla renal y síndrome nefrótico severo refractario al tratamiento con múltiples inmunosupresores, orientó hacia la posibilidad diagnóstica de deficiencia completa de la enzima lecitin-colesterol aciltransferasa; se solicitó secuenciación completa del gen LCAT; el estudio identificó la variante homocigota c.368G > C que correspon de al cambio de una guanina por una citosina en la posición 368 del ADN codificante; esta variante genera el cambio missense de una arginina por una prolina en el aminoácido 123. Posteriormente se realizó biopsia renal que evidenció engrosamiento de la membrana basal capilar y matriz mesangial por presencia de múltiples vacuolas de lípidos. Los resultados del estudio molecular y los hallazgos de la biopsia renal permitieron confirmar el diagnóstico.
Discusión
La deficiencia de LCAT es una enfermedad autosómica recesiva rara, se estima una prevalencia a nivel mundial de 1/1 000 000 habitantes. Hasta el momento se han identificado 60 casos aislados y 70 familias con deficiencia completa o parcial de LCAT. La enfermedad es causada por una muta ción en el gen que codifica para la enzima LCAT, localizado en la región cromosómica 16q22.1. Esta enzima tiene un papel fundamental en la vía metabólica del transporte reverso del colesterol, transformando la HDL inmadura discoidal a una HDL esférica madura por esterificación de colesterol libre en éster de colesterol. La mutación puede ser homo cigota, como en el presente caso, y generar una deficiencia completa de la enzima (enfermedad de Norum), o puede ser heterocigota, causando una deficiencia parcial de la enzima en cuyo caso la forma de presentación es diferente y se le conoce como la enfermedad de los ojos de pescado 1.
The Human Gene Mutation Database (HGMD) ha des crito 102 variantes del gen LCAT relevantes funcionalmente y 77 mutaciones puntuales, dentro de las cuales se incluye la mutación reportada en el presente caso, una mutación homocigota c.368G > C que causa un cambio missense p.Arg123Pro; la cual tiene una frecuencia poblacional muy baja (< 0,0001 gnomAD), mutación no reportada en la lite ratura disponible en Latinoamérica 1.
Clínicamente, la alteración en el metabolismo de las lipoproteínas se manifiesta con HDL marcadamente bajo (< 10 mg/dL), LDL disminuido por el catabolismo acelerado, mientras que las VLDL, el colesterol libre y los triglicéridos están elevados 2. En este caso, el paciente desde los 22 años fue diagnosticado con hipertrigliceridemia severa y recibió manejo con fibratos. Presentaba además HDL y LDL marcadamente bajos, alteraciones que junto a las opacidades corneales difusas (que constituyen el signo más temprano y notorio de la enfermedad y que inicialmente no fueron consideradas relevantes), son características de deficiencia de LCAT 2.
El compromiso renal se manifestó desde la adolescencia como un síndrome nefrótico, se realizó diagnóstico inicial de glomeruloesclerosis focal y segmentaria por los hallazgos en la primera biopsia renal, estudio en el que no fueron evidentes las características histopatológicas típicas de la deficiencia de LCAT, que incluyen en la microscopía de luz el engrasa miento o duplicación de la membrana basal glomerular, con importante proliferación mesangial, y presencia de depósitos de lípidos en la microscopia electrónica, los cuales consisten en pequeñas partículas oscuras de aspecto granular, irregular y electroluscentes localizadas principalmente en los espacios subepitelial, subendotelial, intramembranoso y mesangial 3,4. La ausencia de estos hallazgos probablemente contribuyó a un retraso en el diagnóstico dado que se interpretó como una enfermedad glomerular primaria, por lo que recibió múltiples esquemas de tratamiento inmunosupresor, sin lograr remisión de la enfermedad.
En la literatura reportada de síndrome nefrótico por deficiencia familiar de LCAT, el diagnóstico se realizó con base en las características típicas en la biopsia renal y fue confirmado posteriormente mediante secuenciación genética. Aunque en ninguno se reconoce la GSFS como un patrón de lesión glomerular inicial, Moorhead et al en 1983 reportan el caso de un paciente de nueve años con déficit parcial de LCAT con síndrome nefrótico y GSFS en la biopsia renal sin depósito de lípidos, en este caso el diagnóstico fue realizado por medición directa de la activi dad de la enzima 5,6. El hallazgo de un patrón de GSFS en déficit de LCAT, es de aparición tardía en el curso de la enfermedad y es secundario a una acumulación progresiva de células espumosas vacuoladas que contienen lípidos en el mesangio y la membrana basal glomerular que inducen inflamación y fibrosis 7,8.
Se ha demostrado que la lipoproteína x [Lp(x)] una lipoproteína anormal rica en colesterol libre y fosfolípidos, además de generar lesión por acumulación renal, también es citotóxica y proinflamatoria, estimulando la infiltración de monocitos vía expresión de la proteína quimio-atrayente de monocitos 1 (MCP-1) y aumento en la actividad del factor nuclear kappa B (NF-kB) en las células mesangiales gene rando una respuesta inflamatoria que juega un rol importante en la glomeruloesclerosis 9.
La anemia moderada, normocítica normocrómica no era explicada por la enfermedad renal y se atribuye al depósito de colesterol libre y fosfatidilcolina en la membrana de los glóbulos rojos, lo que acorta su vida media y causa una anemia hemolítica que característicamente es de bajo grado y puede pasar desapercibida. La anemia estaba asociada a esplenomegalia generada por hiperplasia del sistema retículo endotelial que resulta de la destrucción incrementada de eritrocitos defectuosos 10. Se destaca que los estudios de medula ósea fueron normales.
A la fecha no hay un tratamiento específico disponible, la mayoría de los pacientes requieren inicio de terapia de soporte renal y los que logran acceder a un trasplante se ha reportado recurrencia de la enfermedad en el injerto 7. Se está desarrollando la enzima LCAT recombinante humana (rhLCAT) para el tratamiento de la deficiencia completa de la enzima. Los primeros estudios han demostrado normali zación del perfil lipídico, reversión de la anemia, mejoría en la función renal y transformación de las Lp-X en partículas similares a HDL 7.
Conclusión
Para realizar el diagnóstico de deficiencia de LCAT es necesario un alto índice de sospecha al momento de inter pretar las manifestaciones clínicas y las pruebas diagnósticas en las que se incluye la biopsia renal. La confirmación debe realizarse siempre con la secuenciación del gen codificante de LCAT. Es importante realizar consejería genética familiar con el fin de establecer medidas dirigidas a disminuir el ries go de progresión de la enfermedad renal, evitar la realización de estudios innecesarios y esquemas de tratamientos que por la fisiopatología de la enfermedad resultan inefectivos y potencialmente tóxicos.
REFERENCIAS
1. Carmo R, Castro-Ferreira I, Oliveira JP. Lecithin-cholesterol acyltransferase deficiency: a review for clinical nephrologists. Portuguese Journal of Nephrology & Hypertension. 2017;31(4):286-92. [ Links ]
2. Ahsan L, Ossoli AF, Freeman L, Vaisman B, Amar MJ, Shamburek RD, et al. Role of lecithin: cholesterol acyltransferase in HDL metabolism and athero sclerosis. The HDL Handbook: Elsevier; 2014. p. 159-94. https://doi.org/10.3389/fcvm.2020.00065 [ Links ]
3. Najafian B, Lusco MA, Finn LS, Alpers CE, Fogo AB. AJKD Atlas of Renal Pathology: Lecithin-Cholesterol Acyltransferase (LCAT) Deficiency. Ameri can Journal of Kidney Diseases. 2017;70(1):e5-e6. https://doi.org/10.1053/j.ajkd.2015.07.006 [ Links ]
4. Deltas C, Savva I, Voskarides K, Papazachariou L, Pierides A. Carriers of autosomal recessive Alport syndrome with thin basement membrane nephropa-thy presenting as focal segmental glomerulosclerosis in later life. Nephron. 2015;130(4):271-80. https://doi.org/10.1159/000435789 [ Links ]
5. De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC. Differentiating primary, genetic, and secondary FSGS in adults: a clinicopathologic approach. Journal of the American Society of Nephrology. 2018;29(3):759-74. https://doi.org/10.1681/ASN.2017090958 [ Links ]
6. Moorhead J, El-Nahas M, Harry D, Persaud J, Mayne K, Chan M, et al. Focal glomerular sclerosis and nephrotic syndrome with partial lecithin: choles terol acyltransferase deficiency and discoidal high density lipoprotein in plasma and urine. The Lancet. 1983;321(8330):936-7. https://doi.org/10.1016/S0140-6736(83)91368-5 [ Links ]
7. Ossoli A, Neufeld EB, Thacker SG, Vaisman B, Pryor M, Freeman LA, et al. Lipoprotein X causes renal disease in LCAT deficiency. PLoS One. 2016;11(2). https://doi.org/10.1371/journal.pone.0150083 [ Links ]
8. Wahl P, Ducasa GM, Fornoni A. Systemic and renal lipids in kidney disease development and progression. American Journal of Physiology-Renal Physiology. 2016;310(6):F433-F45. https://doi.org/10.1152/ajprenal.00375.2015 [ Links ]
9. Lynn EG, Siow YL, Frohlich J, Cheung GT, Karmin O. Lipoprotein-X stimulates monocyte chemoattractant protein-1 expression in mesangial cells via nuclear factor-xB. Kidney international. 2001;60(2):520-32. https://doi.org/10.1046/j.1523-1755.2001.060002520.x [ Links ]
10. Hirashio S, Ueno T, Naito T, Masaki T. Characteristic kidney pathology, gene abnormality and treatments in LCAT deficiency. Clinical and experimental nephrology. 2014;18(2):189-93. https://doi.org/10.1007/s10157-013-0895-4 [ Links ]
Abreviaturas LCAT: lecitin-colesterol aciltransferasa; HDL: colesterol de alta densidad; LDL: colesterol de baja densidad; GSFS: glomeruloesclerosis focal y segmentaria; HGMD: The Human Gene Mutation Database; VLDL: colesterol de muy baja densidad; Lp(x): lipoproteína x; MCP-1: proteína quimio-atrayente de monocitos 1; NF-kB: factor nuclear kappa B; rhLCAT: enzima LCAT recombinante humana
Recibido: 12 de Enero de 2022; Aprobado: 03 de Febrero de 2022
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
