











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroduction
Thrombotic thrombocytopenic purpura (TTP) is an acute, life-threating disease characterized by microangiopathic he molytic anemia, thrombocytopenia and organ ischemia due to platelet-rich microthrombi, which was first described by Eli Moschcowitz in 1925 1. It is caused by autoantibodies directed against a disintegrin and metalloproteinase with thrombospondin type 1 motifs, member 13 (ADAMTS13), responsible for fragmenting von Willebrand factor. This enzyme deficiency leads to platelet consumption in the platelet/von Willebrand aggregates, causing microvascular thrombosis, and fragmenting and destroying erythrocytes as they pass through the microcirculation. Tissue ischemia and multiple organ dysfunction resulting from microvascular obstruction may lead to thromboembolic events and death. The prevalence in the United States and Europe has been reported to range from 13 to 21 cases per million people, and its incidence from 1.5 to 6 per million people, while its frequency is unknown in Colombia 2,3.
In adults, TTP is hereditary in only 5% of cases, due to mutations in the gene which codes for ADAMTS13 protease, located on chromosome 9. More often, it is acquired (95% of cases), either idiopathic or related to systemic disease that leads to the formation of autoantibodies which increase ADAMTS13 clearance or inhibit its function 4. The risk factors described include recent exposure (within the last two to six weeks) to medications (gemcitabine, oxaliplatin, vincristine, proteosome inhibitors, and calcineurin inhibi tors, among others) and the loss of immune tolerance after exposure to microorganisms with homologous peptides (HIV, hepatitis C virus, S.aureus)5. Diagnostic confirma tion requires showing decreased ADAMTS13 activity and the presence of inhibitors; however, these are not easily available in our setting and results may take one to two weeks. Therefore, based on clinical judgement and the use of factors to predict ADAMTS13 deficit, like the PLASMIC scale, emergent treatment is begun with plasma exchange while awaiting diagnostic confirmation in patients with an intermediate to high likelihood of having the disease, along with immunosuppressant therapy based on glucocorticoids and, sometimes, rituximab. Rituximab aims at avoiding relapses but is limited by the fact that it is not approved by the Instituto Nacional de Vigilancia de Medicamentos y Alimentos (INVIMA) [National Institute for Medication and Food Surveillance] for this condition. The addition of caplacizumab is also recommended, a medication which has recently become available in Colombia, but whose cost-effectiveness has been questioned due to its high cost and low impact on recurrence 6-8.
Previous studies in Colombia are limited to case reports; the series with the most patients had nine, collected from 1992-1998; however, patients with unconfirmed hemolytic-uremic syndrome were included due to the unavailability of laboratory tests at that time 9,10. Given the limitations mentioned, both for diagnosis and treatment, we seek to contribute to the knowledge of the TTP spectrum in our setting, with the description of clinical and laboratory char acteristics, treatment strategies used, frequency of related complications, treatment response markers and in-hospital patient mortality, to help propose strategies aimed at early disease recognition, the development of diagnostic skills and the availability of therapeutic resources to impact the course of this disease which, although rare, is potentially fatal.
Materials and methods
This was an observational, descriptive, retrospective case series study. Patients over the age of 15, of both sexes, who were treated at Hospital San Vicente Fundación (Medellín site) from January 2012 through December 2021 for diagnoses related to thrombotic thrombocytopenic purpura according to the International Classification of Diseases 10th edition (ICD-10): thrombotic microangiopathy (M311) and other non-thrombocytopenic purpuras (D692), were se lected. A search was also done of the blood bank records of patients who required plasma exchange therapy with a diag nosis of TTP, available since 2018. Patients with a diagnosis of TTP in the clinical chart were included, characterized by nonimmune (Coombs negative) hemolytic anemia associated with schistocytes in peripheral blood and thrombocytopenia, with presumed ischemic organ damage, ADAMTS13 levels less than 20% prior to plasma exchange, or higher levels if drawn after plasma exchange, ruling out other potential causes. This was double-checked by the principal investi gator to corroborate the recorded information, and patients diagnosed with catastrophic antiphospholipid syndrome, hemolytic-uremic syndrome and complement-mediated thrombotic microangiopathy were excluded, according to the final diagnosis in the clinical chart and ADAMTS13 levels higher than 20% prior to beginning treatment. Those with complementary studies that did not document an as sociated disease described in the literature were classified as idiopathic TTP. Demographic, clinical and laboratory vari ables, along with treatment plans and treatment responses were described, comparing patients with idiopathic TTP vs. non-idiopathic TTP. Clinical response was defined as platelet counts greater than 150 x 10 9/L and lactate dehy drogenase (LDH) levels less than 1.5 times the upper limit of normal at any time, with a sustained response when no plasma exchanges were required for 30 days. Meanwhile, refractoriness was defined as no significant increase in the platelet count (remaining lower than 50 x 10 9/L) after the first five days of plasma exchanges or clinical deterioration on laboratory tests during daily plasma exchange treatment, and exacerbation referred to a reduction in the platelet count and elevated LDH with no other cause within 30 days of plasma exchange suspension.
Statistical calculations were done using the SPSS (IBM) program, version 28.01.01. The study was considered low-risk, and was approved by the ethics and research committee at Hospital San Vicente Fundación.
Statistical analysis
The distribution of absolute and relative frequencies in each category were determined for qualitative variables, and measures of central tendency, like the mean with its standard deviation or median with its interquartile range, were used for quantitative variables, according to their distribution based on the Shapiro-Wilk test. The Chi2 test was used to compare two qualitative variables (or Fisher's exact test when the 2 x 2 table contained values less than five), and Student's t or the Mann-Whitney U test to compare two quantitative or one qualitative and one quantitative variable, according to the distribution of the data, with differences considered statistically significant if p<0.05.
Results
A total of 509 patients were collected based on the ICD-10 codes; their charts were reviewed, with diagnoses like iso lated cerebral microangiopathy and non-microangiopathic thrombocytopenia not included. Subsequently, 65 patients with a diagnosis of thrombotic microangiopathy were included, 19 of whom were ultimately classified as TTP; those with an initial suspicion and ADAMTS13 levels prior to treatment greater than 20%, those with hemolytic-uremic syndrome with microbiological isolation, and those with complement-mediated thrombotic microangiopathy and catastrophic antiphospholipid syndrome were excluded (Figure 1).
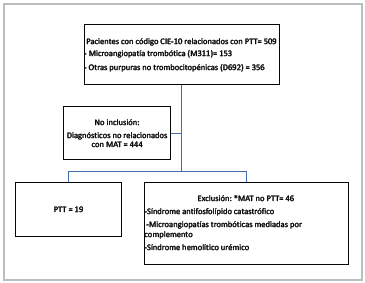
Figure 1 Selection flow chart. Based on the final diagnosis in the medical chart and ADAMTS13 levels under 20% prior to beginning treatment.
Within the demographic characteristics, women were affected more often, at 84.2%, with a 5:1 male:female ratio, a median age of 29 years with an interquartile range (IQR) of 23-49 years, and 52.6% were from Medellín. Altogether, 84.2% were classified as having a first episode, 68.4% had an associated illness, mostly (76%) SLE, there was one TTP relapse related to an 18-week pregnancy, and no other associ ated diseases like malignant neoplasms, medications, infec tion or transplants (solid organ or bone marrow) were found. The most common clinical manifestations were neurological disorders in 73.6% (headache and/or focal deficits in 47.3%), gastrointestinal 5.6% (abdominal pain in 47.3%), fever in 47.3%, and bleeding in 42.1% (88.8% mucocutaneous); 15.7% had an ischemic stroke, 5.2% had the classic pentad (fever, thrombocytopenia, kidney injury, encephalopathy, hemolysis) and 68.4% had kidney involvement, with the latter being more frequent in SLE-associated TTP than in idiopathic TTP (100 vs. 33%; p=0.03) (Table 1, Figure 2).
Table 1 Clinical signs and symptoms according to the systems affected.
| Clinical signs and symptoms | Frequency (%) |
|---|---|
| Fever | 10/19 (47.3) |
| Infection | 3/19 (15.7) |
| Severe HTN on debut | 4/19 (21) |
| Kidney involvement | 13/19 (68.4) |
| *Acute kidney injury | 6/19 (31.5) |
| *Sub-nephrotic proteinuria | 1/19 (5.2) |
| *Nephritic and/or nephrotic syndrome | 6/19 (31.5) |
| Neurological | 14/19 (73.6) |
| *Headache and/or focal deficit | 9/19 (47.3) |
| *Altered consciousness | 2/19 (10.5) |
| *Seizures associated with a headache and/or focalization | 3/19 (15.7) |
| Gastrointestinal | 10/19 (52.6) |
| *Abdominal pain with or without vomiting | 9/19 (47.3) |
| *Diarrhea with or without vomiting | 1/19 (5.2) |
| Bleeding | 9/19 (47.3) |
| *Mucocutaneous (gingival bleeding, purpura) *Melenas (subjective and self-limited) | 8/19 (42.1) 1/19 (5.2) |
| Pulmonary embolism | 1/19 (5.2) |
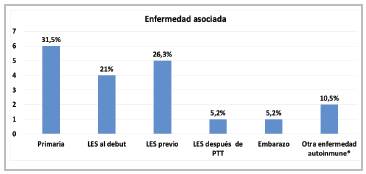
Figure 2 Conditions associated with TTP * Other autoimmune diseases: Sjögren' syndrome and ankylosing spondylitis (autoinflammattory).
The laboratory findings were remarkable for the pres ence of normocytic anemia with a median hemoglobin of 7.7 gr/dL ± 1.7 gr/dL, and a median platelet count of 12 x 109 /L with an IQR of 8-29 x 109 /L. Regarding signs of hemolysis, the reticulocyte production index was 2.6 ± 1.7, with marked LDH elevation (1,509 IU/L ± 862 IU/L) and mild hyperbilirubinemia with a total bilirubin of 2.9 gr/dL (IQR: 1.6-4.1 gr/dL); in addition, haptoglobin was low in all those in whom it was measured (4/4). As far as the tests ordered for the differential diagnosis, none of the patients had low fibrinogen levels, while all those in whom D-dimer was measured had high levels, and none of them had pro longed coagulation times. Antinuclear antibodies (ANAs) were positive in 63.1% of all the patients, with no difference between those with an idiopathic vs. non-idiopathic etiol ogy (p=0.089), and the most frequent patterns were fine and coarse speckled (21%), homogenous (15.7%), and mixed cytoplasmic and homogenous or cytoplasmic and speckled (15.7%), all with high titers ranging from 1:160 to 1:2,560. Additional studies showed hypocomplementemia in 47.3% (77.7% with SLE and 22.3% with idiopathic TTP); all those classified as idiopathic in whom anti-Ro and anti-DNA were measured were negative, while 100% of those who were anti-DNA positive had SLE (p=0.01) (Tables 2 and 3).
Table 2 Laboratory tests for quantitative variables.
| Laboratory tests | Median (interquartile range) | Mean ± standard deviation |
|---|---|---|
| Hemoglobin* | 7.8 gr/dL (6.9-8.8) | 7.7 gr/dL ± 1.7 |
| Hematocrit | 22% (20 - 25.6) | 2.5% ± 5% |
| Mean corpuscular volume * | 87 fL (82-93) | 87.9 ± 7.9 |
| Platelet count | 12 x 109 /L (8-29) | 25 x 109 /L ± 27 |
| Neutrophil count * | 7.4 x 109 /L (4.7-8.2) | 7.8 x 109 /L ± 4.8 |
| Uncorrected reticulocyte percentage * | 6. % (4-144) | 10.1% ± 6.3 |
| Reticulocyte production index * | 2.6 (4.7-8.2) | 2.6 ± 1.7 |
| Total bilirubin | 2.9 mg/dL (1.6- 4.13) | 2.06 mg/dL ± 3.6 |
| Indirect bilirubin | 1.5 mg/dL (0.9-2.4) | 2.35 mg/dL ± 2.9 |
| Lactate dehydrogenase* | 1,428 IU/L (835-2,128) | 1,509 IU/L ± 862 |
| Urea nitrogen | 27 mg/dL (20-34) | 31.3 mg/dL ± 16.3 |
| Creatinine* | 0.9 mg/dL (0.7-1.3) | 1.05 mg/dL ± 0.38 |
| * Determined to have a normal distribution based on the Shapiro-Wilk test. |
Table 3 Complementary laboratory tests.
| Laboratory tests | Positive | Negative | Not measured % |
|---|---|---|---|
| Low ADAMTS13 * | 6 (26.3) | 2 (10.5) | 11 (57.8) + |
| Antinuclear antibodies | 12 (63.1) | 6 (31.5) | 1 (5.2)*** |
| Anti-DNA | 6(31.58) | 11 (57.8) | 2 (10.5) |
| Anti-Ro | 6(31.58) | 8 (42.11) | 5 (26.3) |
| Reduced complement levels | 9 (47.3) | 10 (52.6) | Not applicable |
| Antiphospholipid antibodies | 4(21)** | 14 (73.6) | 1 (5.2) |
* 6 positive patients distributed as follows: 2= idiopathic with levels <10 IU/mL, 1 = idiopathic with levels from 10-20 IU/mL and 3= SLE-associated with levels <10 UI/mL
**Only positive for confirmatory lupus anticoagulant; the rest of the antibodies were negative.
***Not measured during the TTP episode; however, had a history of positive ANAs and SLE.
+ The only patient classified as intermediate risk by PLASMIC was in the unmeasured ADAMTS13 group.
The average duration of symptoms at diagnosis was 10.4 ± 5.9 days; 94.7% were classified as high probability using the PLASMIC scale and 100% using the French scale. ADAMTS13 levels were measured in eight patients (42.1%), five of whom had levels lower than 10 IU/mL (two patients had the test three to ten days after beginning plasma exchange) one had levels between 10 and 20 IU/mL prior to beginning treatment, and two patients had levels greater than 20 IU/mL measured 24 hours after beginning plasma exchange.
Regarding treatment, all patients received plasma exchange and glucocorticoids, combined with cyclophosphamide in 42.1% in the context of SLE-associated TTP; rituximab was ordered for 31.5% (administered to 50% of idiopathic cases), with no difference in those with SLE-TTP according to the treatment scheme (p=0.08). Plasma exchange was begun a median of 1.3 days (IQR: 0-3 days) after TTP diagnosis, with an average total number of plasma exchange sessions of 12.3 ± 9.8, achieving a clinical response within an average of 9.6 ± 8.2 days, with no difference in the number of plasma exchange sessions (p=0.6) or time elapsed to achieve a clinical response (p=0.72) in those classified as having SLE-associated TTP. Clinical response was documented in 78.9%, 31.5% were refractory, 36.8% had an exacerbation, and 15.7% relapsed.
Treatment complications included bacterial infection due to bacteremia in 31.5% (only one related to the catheter), volume overload in 10.5%, and both plasmapheresis-associated hypo calcemia and an allergic reaction to the transfusion in 5.2%. Furthermore, one patient had cytomegalovirus colitis and angioinvasive pulmonary aspergillosis in the context of SLE patients who received three immunosuppression treatments. Finally, 26.3% died in the hospital, with the characteristics of all having died during a first episode of TTP, 80% being refractory and none of them having received rituximab, with no difference in mortality according to the idiopathic or non-idiopathic TTP etiology (p=1.00) (Figure 3, Tables 4 and 5).
Table 4 Response to treatment and complications.
| Treatment outcomes | Frequency (%) |
|---|---|
| Clinical response | 15/19 (78.9) |
| Clinical remission | 11/19 (57.8) |
| Refractoriness | 6/19 (31.5) |
| Exacerbation | 7/19 (36.8) |
| Clinical relapse | 3/19 (15.7) |
| Adverse effects from immunosuppression | 8/19 (42.1) |
| Complications related to plasma exchange | 6/19 (31.5) |
| In-hospital death | 6/19 (26.3) |
Table 5 Clinical, laboratory and treatment characteristics in idiopathic vs. non-idiopathic TTP.
| Characteristics | Idiopathic TTP | Non-idiopathic TTP | P |
|---|---|---|---|
| Kidney involvement | 2/6 (33.3%) | 11/13 (84.6%) | 0.04* |
| Neurological involvement | 6/6 (100%) | 8/13 (61.5%) | 0.12 |
| Gastrointestinal symptoms | 5/6 (83.3%) | 5/13 (38.5%) | 0.14 |
| Fever | 5/6 (83.3%) | 4/13 (30.8%) | 0.05 |
| Platelet count less than 20 x 10 9/L | 6/6 (100%) | 7/13 (53.8%) | 0.10 |
| Hemoglobin | 7.5 gr/dL | 7.7 gr/dL | 0.78 |
| LDH* | 2,164 IU/L | 1,207 IU/L | 0.02** |
| Uncorrected reticulocytes | 13.5% | 8.5% | 0.10 |
| Creatinine | 1.06 mg/dL | 1.05 mg/dL | 0.93 |
| Total bilirubin | 4.4 mg/dL | 2.1 mg/dL | 0.05 |
| Reticulocytes | 13.5% | 8.5% | 0.10 |
| ANA | 2/6 (33.3%) | 10/13 (76.9%) | 0.12 |
| Anti-Ro | 0/6 (0 %) | 6/13 (46.2%) | 0.10 |
| Anti-DNA | 0/6 (0%) | 6/13 (46.2%) | 0.10 |
| Clinical response | 4/6 (66.6%) | 11/13 (84.6%) | 0.55 |
| Exacerbation | 3/6 (50%) | 4/13 (30.7%) | 0.61 |
| Refractory | 3/6 (50%) | 3/13 (23%) | 0.32 |
| In-hospital death | 2/6 (3.3%) | 3/10 (30%) | 1.00 |
| *p<0.05 on Fisher's exact test **p<0.05 on Student's t test |
Discussion
Our results show that TTP most often affects young women and is more frequently associated with another disease than idiopathic. In our series, compared with the largest TTP registry reported by the French referral center for thrombotic microangiopathies, the median age was lower (29 vs. 43 years), with a higher predominance of women (84.2 vs. 68%), and the idiopathic origin was more frequent (68.4 vs. 51%), as was the presence of as sociated autoimmune disease (63.1 vs. 14%) 11. Some autoimmune diseases have been reported to precede it by up to 15 years, be simultaneous or occur during follow up in up to 25.9% within 12 years 11,12. In the current study, SLE was documented in a patient after four years of follow up after the TTP debut, although he initially had a positive ANA with a speckled pattern and titers at 1:320, with negative anti-DNA and anti-Ro. However, it is no table that in SLE-associated TTP vs. idiopathic TTP there was a higher frequency of anti-Ro positivity (50 vs. 11%; p = 0.14) and anti-DNA positivity (60 vs. 0%; p=0.01), which agrees with the predictors described for SLE, such as anti-double-stranded DNA, with a hazard ratio (HR) of 4.98 (p=<001), and anti-Ro (HR 9.98; p=<001) 11,12.
Within the evaluation process, the etiology must be consid ered - hereditary in patients under the age of 25 and when low ADAMTS13 levels are associated with a lack of inhibitors, but this must be confirmed with the detection of the biallelic mutation of the ADAMTS13 gene, which was not done in any of our patients. We reported one case of TTP during the second trimester of pregnancy; the importance of this case lies in the fact that it is a condition described in 12-25% of adult TTPs, generally in women's first pregnancy (76%), who more often have a hereditary cause (24-66%). In these cases, preeclampsia with signs of severity and HELLP syndrome should be included in the differential diagnosis; these should especially be considered if there is clinical resolution within 72 hours of delivery, there is an LDH/AST ratio less than 10 and they occur after the first trimester of pregnancy 13-16.
In this study, there was a notably high occurrence of neurological disorders, even more than in the French regis try (73.6 vs. 61%), as well as digestive symptoms (52.6 vs. 35%), more strokes (15.7 vs. 8%) and fever (47.3 vs. 40%) 11. The laboratory findings showed similar values in the median hematocrit (22 vs. 21 %) and platelet count (12 x 109 /L vs. 10 x 109 /L), with a high frequency of counts over 30 x 109 /L (20% vs. 4%), probably related to SLE-associated TTP (60 vs. 0%; p=0.01) 11,17. Characteristics reported to be associated with idiopathic TTP include digestive symptoms, with an odds ratio (OR) of 19.61, platelets under 20,000/L (OR: 3.18) and anti-ADAMTS13 IgG antibodies (OR: 3.51); however, in our study, lack of kidney involvement and greater LDH elevation were variables related to idiopathic TTP; neurological involvement, fever, platelet counts under 20 x 10 9/L and higher total bilirubin were also more frequent in primary TTP without a statistically significant difference. The strength of association of these factors must be determined in studies with a larger sample size includ ing a significant proportion of people over the age of 60, who tend to present with atypical neurological symptoms and organ dysfunction without severe cytopenias 11,18.
The diagnosis of TTP is a real challenge, with life-or-death consequences and urgency. It is based on the com bination of microangiopathic hemolytic anemia, thrombocytopenia (usually severe, under 30,000/L) and clinical or laboratory signs of variable ischemic effects on some organ, most often the brain, kidney, heart or gastrointestinal system. Confirmation of low ADAMTS13 levels (< 10) is not avail able for several days, which is not in keeping with the need to make rapid lifesaving treatment decisions. Furthermore, after confirming low ADAMTS13 levels, the presence of anti-ADAMTS13 antibodies must be established in order to determine the immune etiology of the condition and instate targeted treatment. However, these laboratory tools are generally not available, but decisions must be made, which will then be based on clinical judgement. Against this back ground, prediction scales like PLASMIC (92-98% negative predictive value, 99% sensitivity and 57% specificity) or the French scale (85% positive predictive value and 93.3% negative predictive value) become important 6,19. While these scales do not replace clinical judgement, they support early initiation of treatment, considering that the measured variables depend on the degree of laboratory abnormalities, severity of organ dysfunction and age (lower PLASMIC sen sitivity in those over the age of 60, 91.4 vs. 76.9%), although it was recently proposed that their discrimination capacity be improved with the addition of the urine protein/creatinine ratio less than 1.2 gr/gr, with an area under the curve (AUC) of 0.76 vs. 0.65 (P=0.003) 20-22. In our study, only one patient was classified by PLASMIC as intermediate prob ability; however, this case would be reclassified as high if the additional proposed variable of lack of proteinuria were included. As far as complementary laboratory tests, low fibrinogen levels and prolonged coagulation times were absent, which suggests their measurement as a contribution to the differential diagnosis of disseminated intravascular coagulation, keeping in mind, however, that there may be coexistence when TTP is associated with infection or sec ondary to severe organ dysfunction 23.
Within the diagnostic process, it is vitally important to determine the presence of hemolysis along with peripheral blood schistocytes (>1% in a count of 1,000 red blood cells per high-power field). In this study, all patients had some hemolysis marker (reticulocytosis: 68%, indirect hyperbilirubinemia: 84%, reduced haptoglobin: 100%, elevated LDH: 100%), which corroborates the need to evaluate all markers, especially the combination of haptoglobin consumption and increased LDH (92% sensitivity and 90% specificity), and even consider correcting the upper limit of total bilirubin with the patient's hematocrit ratio/45, since an interpreta tion with only some tests may be skewed by other causes like elevated haptoglobin in the context of inflammation or reticulocytopenia due to reduced bone marrow response (infection, vitamin B9/B12 deficit, associated bone marrow disease) 24-26. ADAMTS13 deficit can be confirmed using fluorescence resonance energy transfer, ELISA or chemiluminescence methods, together with measurement of inhibitor levels by ELISA, ideally prior to beginning plasma exchange, or at least within the first two sessions (89% detec tion in the first session and 83% in the second session), being careful of potential false reductions secondary to mixing with ethylenediamine tetraacetic acid (EDTA) which inhibits ADAMTS13 in vitro, marked hyperbilirubinemia causing interference by suppressing fluorescence emission or a false elevation due to an extended incubation period after taking the sample 27-31. In our study, ADAMTS13 levels were measured by fluorometry in only 42.8% and were found to be consumed even beginning 3-10 days after starting plasma exchange, and none of the patients had inhibitors measured. Although antibodies may be negative in 5-25% of cases due to low titers, non-neutralizing inhibitors or inhibitors neutral ized by transfusion ADAMTS13, the need to encourage their joint measurement is emphasized, regardless of the delay in reporting and/or treatment initiation time 32.
Compared with an observational TTP study in Asians with or without SLE, our series had lower ANA positivity (63 vs. 71%), but a higher frequency of titers greater than 1:320 (81 vs. 5.6%), probably related to the high percentage of patients with SLE 11,33. An association with TTP should be suspected early in patients with SLE with a hemoglobin less than 7 gr/dL (OR: 6.81 P=0.026), in the context of lu pus activity within the three months prior to hospitalization (OR: 1.54, p=0.026) and in immune idiopathic TTP, those who have lower platelet counts (7.3 x 109 p=0.005) with more involvement of the central nervous system (100% p=0.003) 34-37. Although less kidney involvement (aver age creatinine of 0.83 mg/dL vs. 1.84 mg/dL; p<0.01) and lower mortality (0 vs. 38.9%, p=0.03) have been reported in SLE-associated TTP, it is uncertain if these factors would be replicated in our population, given the more frequent kidney involvement in non-idiopathic TTP found in this series, compared with previous studies 33.
Currently, treatment is based on three pillars: AD-AMTS13 replacement with antibody removal using plasma exchange, reducing antibody production through immunosuppression obtained with high doses of steroids-rituximab, and blocking platelet adhesion using caplacizumab 2. Through the first two pillars, a major therapeutic milestone was achieved, changing an almost-always fatal disease into one with a 91% survival rate, with exchange volumes described in the literature from 40 cc/kg (1 volume) to 60 cc/kg (1.5 volume) once a day or even twice a day in the event of new-onset neurological deficits or severe refractory thrombocytopenia 38-40. The exchange is done with fresh frozen plasma, although there are reports in which a combination with 50% albumin in the first half, or a com bination with cryoprecipitate-poor plasma could save on the number of units of plasma, with similar efficacy, while withdrawing plasma exchange after achieving a clinical response is controversial, as in most cases it does not appear to change the frequency of exacerbations and is associated with more treatment-related complications 41-44. While the recommendation is to begin plasma exchange within four to eight hours after diagnosis, mortality does not appear to be modified in those who receive the exchange within the first 24 hours, [adjusted HR of 0.63 (0.08-4.83)], a strategy used in most of our patients, with an average recorded time elapsed from diagnosis to beginning plasma exchange of 1.3 days (0-3). However, strategies should continue to be strengthened to accelerate the reaction capacity in ordering and implementing treatment as soon as the probability of a TTP diagnosis is determined 45.
Immunosuppression is aimed at obtaining remission and reducing the risk of relapse, with consequent improvement in mortality rates 46,47. In our country, we need to promote the inclusion of an INVIMA indication for rituximab and thus favorably impact the indicators that are negative com pared to international registries, especially those referring to a higher frequency of refractory cases (31.5 vs. 16.1%), ex acerbations (3.6 vs. 26%) and mortality (26.3 vs. 12.4%) 16,48. The recent inclusion of caplacizumab in the therapeutic arsenal for TTP constitutes an additional strategy. This is a nanobody targeted against the A1 domain of von Willebrand factor which interacts with the platelets' glycoprotein Ib-IX-V receptors and, therefore, reduces platelet adhesion and mi crovascular thrombosis. Clinical studies have found that the use of caplacizumab, compared with standard treatment with plasmapheresis and immunosuppression, shows a slightly faster response (2.6 vs. 2.8 days; p=0.01), lower recurrence (12 vs. 38%; p<0.001), fewer exacerbations (4.5 vs. 20.5%; p=<0.05), less refractoriness (4.5 vs. 14.1%; p=<0.05), and, when begun within the first three days of plasma exchange, fewer exchanges (OR: 7.5; p =<0.05) with a 31% reduction in hospital stay (9.9 vs. 14.4 days), and a 65% reduction in intensive care unit stay (3.4 vs. 9.7 days), although with a higher risk of mild to severe mucocutaneous bleeding 49,50. However, despite its approval by regulatory agencies, its universal use for this disease is controversial, due to, among other things, questions regarding its cost-effectiveness and analyses of clinical trials. These have led to uncertainty regarding its general application to all patients, questioning whether the most critical patients should be selected based on as yet undefined criteria or if subgroups with a greater benefit should be determined, allowing it to be proposed as first line or rescue treatment according to individualized patient selection 6.
This study has limitations inherent to its retrospective design, such as confounding bias without the ability to determine a causal relationship in the findings, as well as a potential selection bias, which, while approximating a real-life setting, cannot assure adequate classification, due to the lack of confirmation with ADAMTS13 activity and antibody titer measurement in all patients. However, one of its strengths is the number of patients and prolonged selec tion time compared to previous studies in Colombia, with this being the study with the largest population reported in our setting, which looks at the main characteristics, rein forces the understanding of how to approach patients with suspected TTP in our clinical practice, and shows the need to propose local strategies to improve clinical outcomes.
Conclusions
Thrombotic thrombocytopenic purpura has a heterogenous clinical presentation, in which neurological and gas trointestinal symptoms predominate. The laboratory results are notable for marked elevation of lactate dehydrogenase with incipient elevation of bilirubin and reticulocytes. Its diagnosis requires ADAMTS13 activity measurement prior to beginning plasma exchange or even after, interpreting the result with caution according to the context, as well as evaluating inhibitor levels. Within the comprehensive ap proach, it is essential to evaluate the presence of systemic lupus erythematosus both in the debut as well as in long-term follow up, especially in those with kidney disease and positive antinuclear antibodies, as well as ensure appropriate follow up of women of reproductive age with a history of TTP (pre-conception and during pregnancy) together with an adequate etiological classification. The high mortality and frequency of refractoriness warrant determining potential causes, promoting strategies for timely diagnosis and widen ing the availability of all treatment pillars.

