










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Química
versión impresa ISSN 0120-2804versión On-line ISSN 2357-3791
Rev.Colomb.Quim. v.37 n.1 Bogotá ene./abr. 2008
APMO: UN PROGRAMA COMPUTACIONAL PARA EL ESTUDIO DE EFECTOS CUÁNTICOS NUCLEARES MEDIANTE LA TEORÍA DEL ORBITAL MOLECULAR ELECTRÓNICO Y NO ELECTRÓNICO
APMO: A COMPUTER PROGRAM BASED ON THE ELECTRONIC AND NON-ELECTRONIC MOLECULAR ORBITAL THEORY FOR STUDIES OF NUCLEAR QUANTUM EFFECTS
APMO: UM PROGRAMA COMPUTACIONAL PARA O ESTUDO DOS EFEITOS QUÂNTICOS NUCLEARES POR MEIO DO TEORIA DO ORBITAL MOLECULAR ELETRÔNICO E NÃO - ELETRÔNICO
Sergio A. González1, Néstor F. Aguirre1, Andrés Reyes1
1 Departamento de Química, Facultad de Ciencias, Universidad Nacional de Colombia, sede Bogotá, Bogotá, Colombia. areyesv@unal.edu.co
Recibido: 27/11/07 – Aceptado: 07/04/08
RESUMEN
Con el fin de estudiar teóricamente fenómenos en donde los núcleos atómicos presentan comportamiento cuántico, hemos desarrollado el paquete computacional APMO (Any-Particle Molecular Orbital). Éste implementa el método de orbitales moleculares nucleares y electrónicos (OMNE) a un nivel de teoría Hartree- Fock (HF), en el que tanto núcleos como electrones se representan como funciones de onda.
Para comprobar la correcta implementación del método se realizaron cálculos de estructura electrónica regular y núcleo- electrónica de las moléculas H2 y LiH. Las componentes de energía calculadas siguen las tendencias y están en el mismo orden de magnitud de cálculos similares reportados en la literatura.
A diferencia de otros paquetes que implementan el método OMNE, el nuestro fue diseñado para estudiar sistemas con cualquier número de especies cuánticas.
Palabras clave: efectos cuánticos nucleares, efecto isotópico, Hartree-Fock, orbitales núcleo-electrónicos, aproximación de Born-Oppenheimer.
ABSTRACT
With the aim of studying phenomena where atomic nuclei have a quantal behavior, we have developed the APMO (Any-Particle Molecular Orbital) software package. This implements the nuclear and electronic molecular orbital approach (NEMO) at a Hartree-Fock level of theory, where both nuclei and electrons are represented as wave functions.
To verify the correct implementation of the method, a number of electronic and nuclear-electronic calculations were carried out on H2 and LiH molecules. The calculated energy components follow the trends and are of the same order of magnitude of similar calculations reported in the literature.
In contrast to other packages that implement the NEMO approach, ours is designed to allow for studying systems with any number of quantum particles.
Key words: Nuclear quantum effects, isotope effect, Hartree-Fock, nuclearelectronic orbitals, Born-Oppenheimer approximation.
RESUMO
Com a finalidade de estudar teoricamente fenômenos cujos núcleos atômicos apresentam comportamento quântico, desenvolvemos o pacote computacional APMO (Any-Particle Molecular Orbital). Este implementa o método de orbitais moleculares nucleares e eletrônicos (OMNE) no nível da teoria Hartree-Fock (HF), onde tanto núcleos como elétrons se apresentam como funções de onda.
Para comprovar a utilização correta do método, se realizaram cálculos da estrutura eletrônica regular e do núcleo eletrônico das moléculas H_2 y LiH. Os componentes de energia calculados seguem as tendências e estão na mesma ordem de magnitude de cálculos similares reportados na literatura.
Diferentemente de outros pacotes que utilizam o método OMNE, o nosso foi desenhado para estudar sistemas de qualquer número de espécies quânticas.
Palabras-chave: Efeitos quânticos nucleares, efeito do isótopo, Hartree- Fock, orbital núcleo-eletrônico, aproximação do Born-Oppenheimer.
INTRODUCCIÓN
La aproximación de Born-Oppenheimer (ABO) es tal vez la más importante de la química cuántica. La aplicación de esta aproximación ha hecho posible el estudio de sistemas que contienen miles de átomos. A pesar de su éxito en muchas áreas de investigación, los métodos basados en la ABO no permiten hacer una descripción correcta de fenómenos en los que se evidencia el comportamiento cuántico de los núcleos atómicos. Entre estos fenómenos tenemos las vibraciones y el tunelamiento nuclear, los efectos de isótopo, entre otros.
Para realizar un estudio teórico de fenómenos donde falla la ABO, como son los efectos cuánticos nucleares (ECN), existen actualmente dos metodologías: la primera consiste en realizar cálculos de estructura electrónica convencional (basados en la ABO) los cuales se corrigen para incorporar los efectos de las masas y las velocidades nucleares. Entre las correcciones más comunes se encuentran la de Bell para el tunelamiento (1), la armónica para la energía de punto cero, la de masa para los efectos de isótopo. La segunda metodología se fundamenta en el uso de métodos que van más allá de la ABO, los cuales permiten calcular directamente los ECN a partir de un cálculo similar a uno de estructura electrónica convencional. Entre los métodos más reconocidos se encuentran el de Orbitales Núcleo- Electrónicos (NEO) (2), el de Orbitales Moleculares de Multicomponentes (MCMO) (3-4) y el de Orbitales Moleculares Nucleares y Electrónicos (NOMO) (5). En estos métodos los núcleos que presentan comportamiento cuántico se tratan al mismo nivel que los electrones, a través de orbitales moleculares expresados en términos de combinaciones lineales de funciones gausianas.
Ya que las implementaciones computacionales de los métodos anteriormente mencionados presentan limitaciones relacionadas con el número de especies no electrónicas soportadas (método NEO) o con la disponibilidad del software (métodos NOMO y MCMO), hemos construido el paquete computacional APMO (Any Particle Molecular Orbitals) (6). Éste nos ha permitido estudiar ECN a un nivel de teoría Hartree-Fock en sistemas que contengan mezclas de partículas cuánticas . Una de las ventajas del paquete APMO es que sigue una filosofía de programación orientada a objetos, la cual permitirá (en proyectos posteriores) realizar fácilmente extensiones del programa a niveles superiores de teoría (DFT, MPn, CI, MCSCF).
En este documento se presentan los detalles técnicos del programa APMO. Para examinar la correcta implementación del método de orbitales núcleoelectrónicos se realizaron cálculos de estructura electrónica y núcleo-electrónica de las moléculas H2 y LiH, los cuales se compararon con resultados reportados en la literatura.
MÉTODOS
Descripción del modelo
En esta sección presentamos brevemente las expresiones generales del método OMNE. Los detalles completos pueden consultarse en los artículos originales (2-4).
En este método, el hamiltoniano de un sistema que contiene mezclas de especies cuánticas y clásicas está dado por (en unidades atómicas)
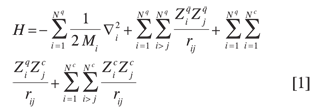
donde el primer término corresponde a la energía cinética de las partículas cuánticas, el segundo término corresponde a las interacciones entre pares de partículas cuánticas con cargas Ziq y Zjq, el tercer término corresponde a las interacciones entre una partícula cuántica de carga Zi q y una clásica con carga Zjc, y el último término representa las interacciones entre partículas clásicas con cargas Zic y Zjc. Nq y Nc son el número de partículas cuánticas y clásicas.
A un nivel de teoría Hartree-Fock, la función de onda total del sistema descrito por el hamiltoniano [1], se puede escribir como el producto de las funciones de onda de las diferentes especies cuánticas:

donde cada función Φi puede ser simétrica o antisimétrica dependiendo de su naturaleza bosónica o fermiónica respectivamente.
La ecuación de Hartree-Fock para una partícula de la especie α tiene la misma forma que en el caso electrónico convencional
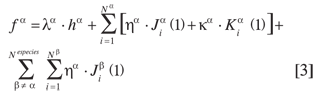
donde el operador efectivo de Fock para una partícula de la especie α es

En la expresión anterior, N(α) es el número de partículas de la especie α; h(α) es el operador de partícula independiente, y J(α) y K(α) son los operadores de coulomb e intercambio respectivamente (2). J(β) es el operador de interacción entre diferentes especies; el parámetro K(α) toma el valor de -1 para una especie fermiónica y +1 para una especie bosónica.
Los orbitales moleculares de la especie α se construyen a partir de una combinación de Nαbf funciones base tipo gausiana (FTG).
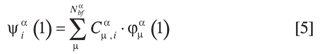
La expresión de la energía total de un sistema con múltiples especies cuánticas es
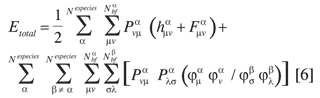
donde se considera una configuración electrónica en capa cerrada, y una configuración para las especies no electrónicas en capa abierta y máximo espín. h(α), F(α) son las representaciones matriciales de los operadores de partícula independiente y de Fock. P(α) es la matriz de densidad.
DESARROLLO COMPUTACIONAL
Diseño. APMO ha sido escrito en C++ y Fortran siguiendo una filosofía de programación orientada a objetos, la cual facilita el mantenimiento y la extensibilidad del código.
El programa utiliza dos bibliotecas, una para realizar procedimientos numéricos y de álgebra lineal (GNU Scientific Library - GSL) (7), y otra para acceder a las bases de datos con información sobre elementos y bases de funciones gausianas (biblioteca EXPAT - XML parser) (8).
En el diseño orientado a objetos, los procedimientos asociados a las ecuaciones [3-6] son extensibles a cualquier especie cuántica: núcleos, electrones, muones, positrones (las dos últimas dentro de un formalismo HF), entre otras.
Uso del programa. El programa se puede utilizar de dos maneras. La primera a nivel de programador, el cual tiene a su disposicion un API (application programming interface), que le permite manipular la estructura del software directamente, además de facultarlo para la fácil adición de nuevos métodos. La segunda a nivel de usuario, el cual puede ejecutarlo de la misma manera que programas de estructura electrónica tales como GAMESS (9) o NWCHEM (10), es decir, por medio de archivos de entrada.
Estructura del programa
La Figura 1 muestra el diagrama de componentes UML simplificado de APMO, en él se muestran sus bloques básicos y las relaciones que existen entre ellos. A continuación se describirá brevemente cada componente.
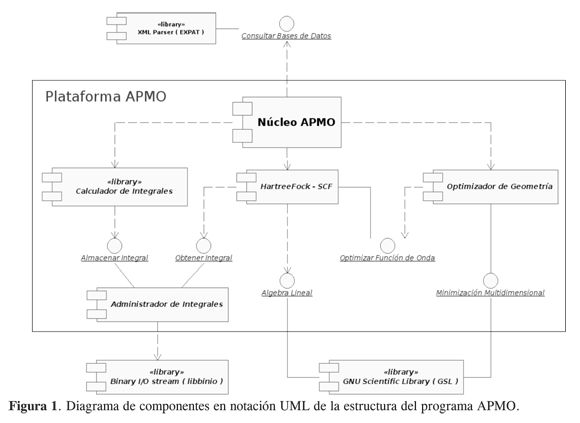
Cálculo y almacenamiento de integrales. El cálculo de integrales (energía cinética, atracción, repulsión, etc.) para funciones base gausianas se realiza empleando los algoritmos recursivos propuestos por Obara y Saika (11). Dado que el cálculo de integrales es el paso de mayor costo computacional, estos algoritmos fueron implementados en Fortran, debido a su alto desempeño en velocidad.
Para el cálculo de integrales se ha implementado un componente (calculador de integrales), que se encarga de calcular y enviar las integrales a una base de datos a través del “Administrador de integrales”; este componente puede almacenar los valores de las integrales en memoria o disco, para el último caso se emplea la librería libinio, que permite almacenar las integrales en forma binaria. Una vez almacenadas quedarán a disposición de cualquier componente del programa.
Consulta a bases de datos. Para consultar bases de datos APMO cuenta con el componente “XML Parser”, éste se encarga de consultar las bases de datos para: partículas (con información: carga, masa, espín, entre otras), elementos (con información: número atómico, lista isótopos, masa nuclear, etc.) y funciones base (con información sobre tamaño de la base, exponentes y coeficientes de contracción), dichas bases de datos residen en archivos escritos en formato XML, y son consultadas a través de la biblioteca EXPAT (8).
Implementación del método ONMERHF. APMO cuenta con un componente que resuelve las ecuaciones de Hartree- Fock [3-6] para cada especie; éstas se resuelven iterativamente mediante un esquema SCF que se detiene una vez la desviación estándar de los elementos de la matriz de densidad para cada especie cuántica alcanza un umbral establecido. El algoritmo variacional del método OMNE adopta un esquema iterativo que realiza la convergencia total de la parte electrónica por cada ciclo SCF de cualquier otra especie no electrónica. Los procedimientos de álgebra lineal requeridos en la solución de las ecuaciones de Fock, hacen uso del componente de álgebra lineal de la biblioteca GSL (7).
Optimización de geometría. Para la optimización de geometría APMO utiliza los métodos numéricos de minimización multidimensional que provee la biblioteca GSL (algoritmos de gradiente conjugado Fletcher-Reeves o Polak-Ribiere, el método cuasi-Newton Broyden-Fletcher- Goldfarb-Shanno y el algoritmo simplex de Nelder y Mead). Por omisión, la implementación utiliza el algoritmo de Broyden-Fletcher-Goldfarb-Shanno, con un criterio de finalización basado en una tolerancia del gradiente de 1E-6 Hartree/ Bohr.
RESULTADOS Y DISCUSIÓN
Con el fin de validar la implementación y probar las capacidades de APMO, se realizaron cálculos HF de estructura electrónica, los cuales se compararon con los obtenidos utilizando el paquete GAMESS (9). Adicionalmente, se realizaron cálculos HF de estructura núcleo-electrónica, los cuales se compararon con cálculos análogos realizados por Tachikawa (12).
Se escogieron como sistemas de prueba las moléculas H2, LiH y H2O, en las cuales los electrones, y los núcleos de hidrógeno y de litio se trataron como especies cuánticas. Para los cálculos de estructura electrónica se utilizaron las bases electrónicas STO-3G y 6-31G. Mientras que los cálculos de estructura núcleoelectrónica emplearon una base STO-3G para la parte electrónica, y una función base 1s para cada núcleo cuántico (con exponentes 22.3, 32.8, 41.0 y 578.785 para el protio, deuterio, tritio y litio (6Li) respectivamente).
En la Tabla 1 se reportan las energías electrónicas obtenidas con APMO, y se comparan con aquéllas obtenidas usando GAMESS. Se observan desviaciones respecto a GAMESS entre 1,2E-6 y 5,5E-10 Hartree. Al analizar las componentes de energía se encuentra que los mayores aportes a la desviación son producidos por las energías repulsivas Vee y las atractivas Vne, las cuales APMO evalúa por métodos de integración diferentes a los usados en GAMESS.
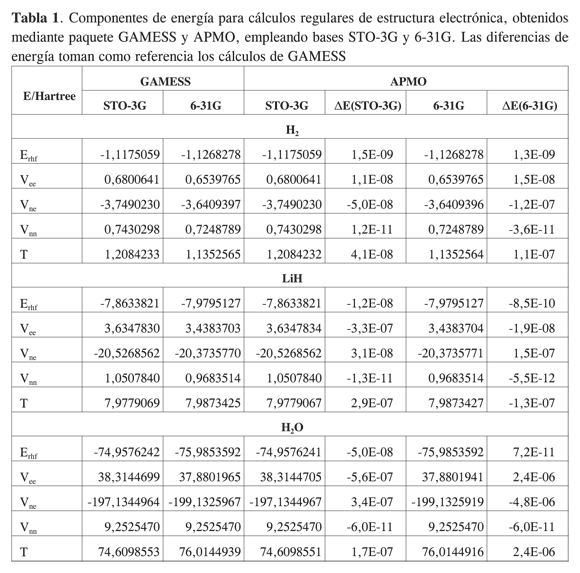
En las Tablas 2 y 3 se registran las componentes de energía calculadas y reportadas para algunos isotopólogos* del hidrógeno molecular H2 y del hidruro de litio LiH. A pesar de que las funciones base del cálculo reportado en la literatura [4s:1s1p] (12) no son las mismas que las utilizadas por APMO [STO-3G:1s], los órdenes de magnitud de cada componente y las tendencias de los resultados concuerdan entre las dos fuentes.
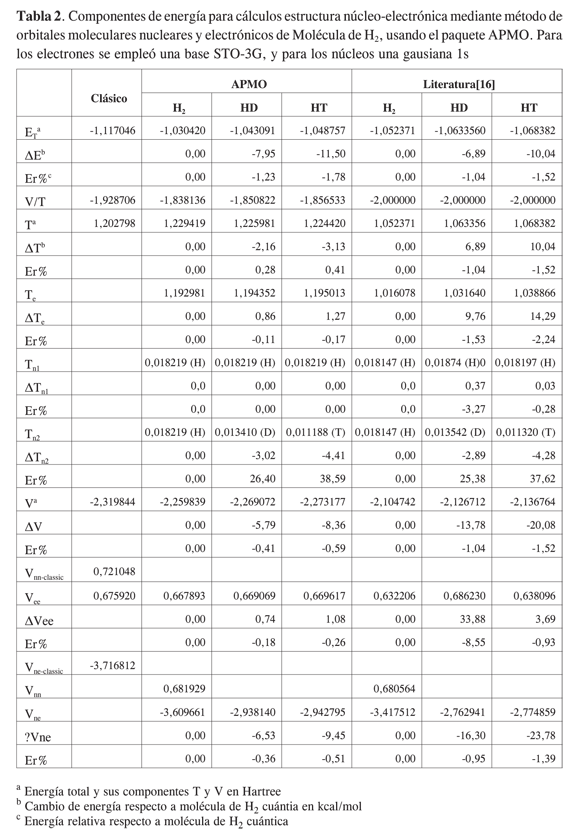
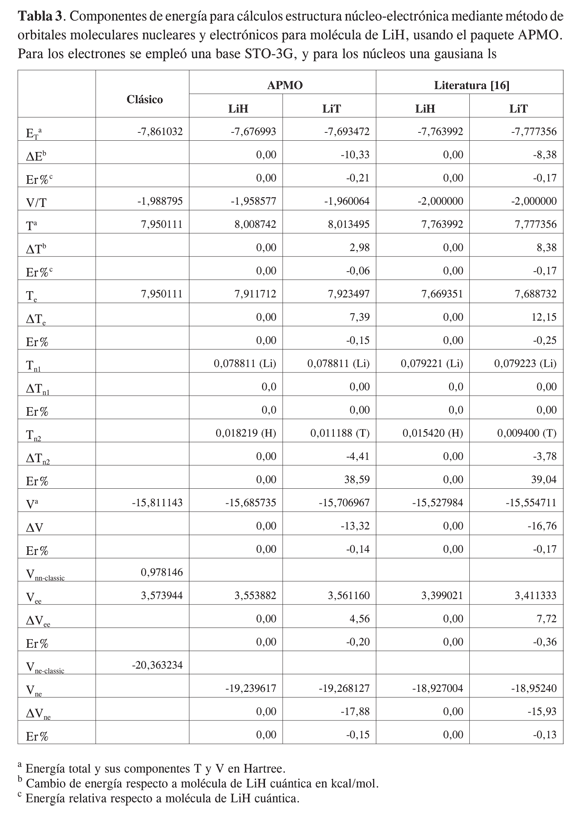
En cuanto a la energía potencial, si bien los cambios globales van en aumento por sustitución con núcleos más pesados, se observan desviaciones en las componentes para cada especie respecto a las tendencias reportadas; así, mientras APMO muestra que la energía potencial electrónica aumenta por sustituciones de protio a tritio, en la literatura el mayor aumento se da por sustitución con deuterio, este comportamiento se debe a los cambios completamente variacionales realizados en las bases de la literatura para la parte electrónica, y que APMO mantiene constantes.
Las variaciones relativas de las energías totales de HD y HT respecto a H2empleando APMO fueron de -1,23 y -1,78%, mientras que las reportadas en la literatura fueron de -1,04 y -1,52% para los mismos sistemas. La variación de la energía del LiD fue de -0,21% respecto al valor del LiH empleando APMO, mientras en la literatura fue -0,17%. Como se observa, las variaciones en energía obtenidas con APMO mantienen las mismas tendencias que las encontradas en la literatura, a pesar de las diferencias en las bases empleadas.
Como se ha mencionado, las diferencias encontradas entre las componentes de energía se atribuyen a las discrepancias entre nuestras funciones base y las utilizadas en la literatura, las cuales se modifican variacionalmente durante el cálculo. Como se observa en las Tablas 2 y 3, los cambios en la base electrónica tienen un impacto mayor en las componentes de energía que los cambios en las bases nucleares. Según Tachikawa (12), las desemejanzas en la base electrónica han sido producto de la naturaleza cuántica de los núcleos, y son mayores cuanto más ligeros son los mismos. Lo anterior muestra la necesidad de diseñar bases electrónicas y nucleares apropiadas para sistemas que contengan núcleos livianos, con el fin de evitar el aumento o la disminución indebido del efecto nuclear.
CONCLUSIONES
Fue posible realizar la implementación de un programa computacional para cálculo de estructura núcleo-electrónica a nivel HF, mediante una filosofía orientada a objetos utilizando los lenguajes de programación C++ y Fortran.
Los resultados obtenidos para cálculos regulares de estructura electrónica son comparables a los encontrados con el programa GAMESS. Los cálculos de estructura núcleo-electrónica obtenidos mediante el método OMNE muestran tendencias y variaciones de energía total, y de sus componentes, acordes con los reportes de la literatura para métodos equivalentes, indicando las posibilidades del programa en el estudio de efectos cuánticos nucleares; sin embargo, debe considerarse el diseño de bases electrónicas y nucleares adecuadas para lograr una correcta descripción del efecto nuclear en sistemas que contengan núcleos ligeros.
El programa APMO será puesto próximamente a disposición de la comunidad científica.
AGRADECIMIENTOS
La presente investigación ha sido financiada por la División de Investigaciones (DIB) de la Universidad Nacional de Colombia, sede Bogotá, proyecto 20101007499, a quienes los autores expresan su agradecimiento.
REFERENCIAS BIBLIOGRÁFICAS
1. Bell, R.P. The tunnel effect in chemistry. London, Chapman and Hall. 1980. [ Links ]
2. Webb, S.P.; Iordanov, T.; Hammes- Schiffer, S. Multiconfigurational nuclear-electronic orbital Approach: Incorporation of Nuclear Quantum Effects in Electronic Structure Calculations. J. Chem. Phys. 2002. 117 (9): 4106-4118. [ Links ]
3. Tachikawa, M.; Mori, K.; Nakai, H.; Iguchi, K.. An Extention of ab initio Molecular Orbital Theory to Nuclear Motion. I. Chem. Phys. Lett. 1998. 290 (4-6): 437-442. [ Links ]
4. Tachikawa, M. Multi-component Molecular Orbital Theory for Electron and Nuclei including manybody effect. Chem. Phys. Lett. 2002. 360 (5-6): 494-500. [ Links ]
5. Nakai, H. Simultaneous Determination of Nuclear and Electronic Wave Functions without Born-Oppenheimer Approximation: Ab Initio NO+MO/HF Theory. Int. J. Quant. Chem. 2002. 56 (6): 511-517. [ Links ]
6. González, S.A.; Aguirre, N.F.; Reyes, A. Theoretical investigation of isotope effects: The Any-Particle Molecular Orbital code. Int. J. Quant. Chem. 2008. En prensa. [ Links ]
7. GSL (GNU Scientific Library) [online]. Boston, USA. Free Software Foundation, Inc. October 2007. [citada 30-10-2007]. Disponible en: http://www.gnu.org/software/gsl/ [ Links ]
8. EXPAT (XML parser library) [online]. [citada 30-10-2007]. Disponible en: http://expat.sourceforge.net/ [ Links ]
9. Schmidt, M. W.; Baldridge, K. K.; Boatz, J. A.; Elbert, S. T.; Gordon, M. S.; Jensen J. J. GAMESS User's Guide. Ames, Iowa: Iowa State University. 2007. [ Links ]
10. Kendall, R.A.; Apra, E.; Bernholdt, D.E.; Bylaska, E.J.; Dupuis, M.; Fann, G.I. High Performance Computational Chemistry: An Overview of NWChem a Distributed Parallel Application. Computer Phys. Comm. 2000. 128 (1-2): 260-283. [ Links ]
11. Obara, S.; Saika, A. Efficient Recursive Computation of Molecular Integrals Over Cartesian Gaussian Functions. J. Chem. Phys. 1987. 84 (7): 3963-3974. [ Links ]
12. Tachikawa, M.; Osamura, Y. Isotope effect of hydrogen and Lithium Hydride molecules. Application of DEMO method and Energy Component Analysis. Theor. Chem. Acc. 2000. 104 (1): 29-39. [ Links ]

