










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804On-line version ISSN 2357-3791
Rev.Colomb.Quim. vol.38 no.1 Bogotá Jan./Apr. 2009
IMPLEMENTACIÓN DEL MÉTODO DEL GRADIENTE ANALÍTICO DE LA ENERGÍA EN LA TEORÍA DEL ORBITAL MOLECULAR NUCLEAR Y ELECTRÓNICO
IMPLEMENTATION OF THE ANALYTICAL ENERGY GRADIENT METHOD IN THE NUCLEAR AND ELECTRONIC MOLECULAR ORBITAL THEORY
EXECUÇÃO DO MÉTODO DE INCLINAÇÃO ANALÍTICO DA ENERGIA NA TEORIA DO ORBITAL MOLECULAR NUCLAER E ELETRÔNICO
Sergio A. González1, Andrés Reyes1
1 Departamento de Química, Facultad de Ciencias, Universidad Nacional de Colombia, sede Bogotá, Bogotá, Colombia. areyesv@unal.eduxo
Recibido: 27/11/08 - Aceptado: 13/04/09RESUMEN
Con el fin optimizar las geometrías promedio de sistemas moleculares utilizando la teoría del orbital molecular nuclear y electrónico (OMNE), se dedujo la expresión para el cálculo del gradiente analítico de la energía a nivel de teoría Har-tree-Fock, para cualquier tipo de especie cuántica. La implementación computacional se realizó dentro del paquete APMO (Any-Particle Molecular Orbital), y con el fin de comprobar la correcta implementación del método se calcularon las moléculas modelo H2,HF y H2O, por medio de métodos numéricos y analíticos. El uso de derivadas analíticas dentro del formalismo OMNE permitirá el cálculo más eficiente de la estructura núcleo-electrónica de sistemas moleculares con el paquete APMO.
Palabras clave: gradiente analítico de la energía, efectos cuánticos nucleares, Hartree-Fock, orbitales nucleares y electrónicos, aproximación de Born-Oppenheimer.
ABSTRACT
In order to optimize the average geometries of molecular systems using the nuclear and electronic molecular orbital theory (NEMO), we have deducted the expression for calculating the analytical gradient of the energy in the Hartree-Fock theory, for any kind quantum specie. The implementation was done within the computational package APMO (Any-Particle Molecular Orbital) and in order to verify the correct implementation of the method, we have calculated the model molecules H2, HF and H2O, with numerical and analytical methods. With the use of analytical derivatives within of the OMME formalism, we will have a more efficient calculation of the nuclear-electronic structure of molecular systems with the APMO package.
Key words: analytical energy gradient, nuclear quantum effects, Hartree-Fock, nuclear-electronic orbitals, Born-Oppenheimer approximation.
RESUMO
Com a finalidade de otimizar as geometrias médias de sistemas moleculares utilizando a teoria do orbital molecular nuclear e eletrônico (OMNE), se deduziu a expressão para o cálculo do gradiente Analítico da energia a nível de Hartree-Fock, para qualquer tipo de espécie quântica. A implementação computacional se realizou dentro do pacote APMO (Any-Particle Molecular Orbital), e com a finalidade de comprovar a correta implementação do método foram calculadas as moléculas modelo H2,HFeH2O, por meio de métodos numéricos e analíticos. O uso de derivadas analíticas dentro do formalismo OMNE vai permitir um cálculo mais eficiente da estrutura núcleo-eletrônica de sistemas moleculares com o pacote AMPO.
Palavras-chave: inclinação analítico da energia, efeitos quânticos nucleares, Hartree-Fock, orbital núcleo-eletrônico, aproximação do Born-Oppenheimer.
INTRODUCCIÓN
Desde un punto de vista matemático, el término gradiente hace referencia a la dirección en la cual una función definida en un espacio n-dimensional alcanza su máxima tasa de cambio. El cálculo de gradientes se hace necesario en problemas donde se desea determinar los puntos esracionarios de una función multidimensional. En química computacional, la determinación de la estructura molecular de mínima energía por medio de métodos teóricos requiere la evaluación del gradiente de la energía respecto a las posiciones nucleares.
El gradiente de la energía puede calcularse por métodos numéricos o analíticos. La evaluación numérica del gradiente se realiza con el método de diferencias finitas. La exactitud de este método depende de la elección del tamaño del paso y del número de puntos con que se aproxima la derivada. En el esquema más simple se requieren al menos dos cálculos de energía por coordenada nuclear, es decir, el cálculo del gradiente escala con 6N (N es el número de centros), lo cual implica un gran costo computacional.
La evaluación analítica del gradiente se realiza a partir de la expresión analítica de la derivada de la energía con respecto a las coordenadas nucleares. El cálculo de gradientes analíticos tiene la ventaja de ser más eficiente y no estar limitado a la elección de un tamaño de paso, cuyo límite infinitesimal puede llevar a problemas numéricos (de punto flotante).
Pulay, en 1969 (1), dedujo la expresión de la primera derivada analítica de la energía electrónica Hartree-Fock (HF), respecto a variables geométricas. Hoy en día el método se ha aplicado a todas las expresiones de energía de los métodos de estructura electrónica disponibles, desde métodos de una sola configuración hasta métodos multiconfiguracionales y semiempíricos (2).
El uso del gradiente analítico también ha sido aplicado a métodos ab initio que van más allá de la aproximación de Born-Oppenheiner (ABO) (3), como el método de orbitales moleculares nucleares y electrónicos (OMNE), que permite obtener de manera simultánea funciones de onda nucleares y electrónicas sin recurrir a la ABO, el cual ha sido desarrollado por varios grupos de investigación (4-8). Sin embargo, las implementaciones computacionales de estos métodos no se encuentran disponibles o están limitadas al número de especies cuánticas. Por esta razón se ha derivado e implementado computacionalmente la expresión general del gradiente analítico para cualquier tipo de partícula cuántica dentro del paquete APMO (9, 10).
En este documento se presentan las expresiones de la energía y de su derivada respecto a las posiciones de los centros de las funciones base nucleares y de las cargas nucleares puntuales, que los autores de esta investigación han deducido a partir de la teoría OMNE/HF para cualquier tipo de partícula cuántica.
Para comprobar la correcta implemen-tación del método de derivadas analíticas dentro del paquete APMO, y su eficiencia respecto al método numérico, se presentan algunas aplicaciones sobre los sistemas moleculares: H2,HFyH2O en donde los electrones y los núcleos de hidrógeno se consideran como partículas cuánticas. Los tiempos de cálculo muestran las ventajas del método de gradiente analítico sobre el método numérico.
ASPECTOS TEÓRICOS
Método de orbitales moleculares nucleares y electrónicos
En publicaciones previas, los autores mostraron las expresiones generales del método OMNE, al igual que algunos detalles de la implementación computacional del mismo (9-10). Una descripción completa del método puede consultarse en los artículos originales (4-8). En el método OMNE, el hamiltoniano de un sistema que contiene mezclas de especies cuánticas y clásicas está dado por (en unidades atómicas):
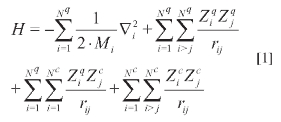
donde el primer término corresponde a la energía cinética de las partículas cuánticas; el segundo término, a las interacciones entre pares de partículas cuánticas con cargas Ziq y Zjq, y el tercer término, a las interacciones entre una partícula cuántica de carga Ziq y una clásica con carga Zjc; el último término representa las interacciones entre partículas clásicas con cargas Zic y Zjc.Los índices Nq y Nc sonel númerodepartícu-las cuánticas y clásicas. Aunnivelde teoría Hartree- Fock, las ecuaciones OMNE/HF pueden obtenerse asumiendo una función de onda total, construida como el producto de funciones de onda, simétricas o anti- simétricas, para las diferentes especies cuánticas, dependiendo de si se trata de bosones o fer-miones, respectivamente.
Las ecuaciones OMNE/HF pueden resolverse variacionalmente, de modo que la expresión de la energía total para un sistema con múltiples especies cuánticas es:
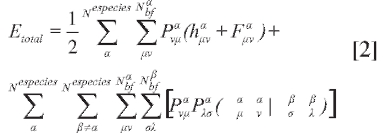
donde se considera una configuración electrónica en capa cerrada y una configuración para las especies no electrónicas, en capa abierta y máximo espín. h(α), F(α) son las representaciones matriciales de los operadores de partícula independiente y de Fock. P(α) es la matriz de densidad.
Cálculo del gradiente analítico de la energía
Para hallar un mínimo de energía de un sistema molecular con el método OMNE, es necesario optimizar los centros de las funciones base de los núcleos y las posiciones de las cargas puntuales. Esto se logra empleando un método de minimización multidimensional que requiere el cálculo del gradiente de la energía. Este último demanda el cálculo de las derivadas de las integrales moleculares presentes en la expresión de la energía [3] (traslapamiento, energía cinética, atracción y repulsión).
La expresión para el gradiente de la energía OMNE/HF, respecto a coordenadas nucleares en términos de los orbitales atómicos, fue deducida a partir de la metodología utilizada en cálculos de estructura electrónica regular (2), la cual se extendió para considerar cualquier tipo y número de especies cuánticas (9). Las expresiones de las derivadas de integrales moleculares fueron obtenidas derivando las expresiones recursivas de Obara y Saika (11) para electrones, y extendiéndolas a todo tipo de partículas cuánticas.
La expresión obtenida para la derivada de la energía total, respecto a una coordenada arbitraria RNX es la siguiente:
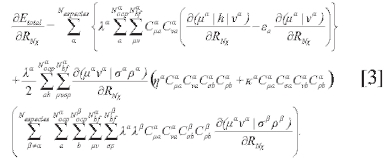
Las constantes λ, η y κ se denominan constantes de acoplamiento y deben ser ajustadas dependiendo del tipo de especie cuántica, tal como se muestra en (9-10). En la ecuación [3] el segundo término en el primer par de sumatorias está relacionado de manera aproximada con la derivada de los coeficientes de combinación de los orbitales atómicos respecto a las coordenadas nucleares (fuerza de la función de onda), mientras los otros términos están relacionados aproximadamente con la fuerza de Hellman-Feynman.
Es bien sabido que la implementation computacional directa de la ecuación [3] es poco eficiente; por esta razón se utilizó un algoritmo análogo al propuesto en (2).
El método de gradiente analítico fue implementado dentro del paquete computacional APMO (9), y acoplado al método de minimización multidimensional cuasi-Newton Broyden-Fletcher-Goldfarb-Shanno tomando el algoritmo de Shano y Phua, el cual se encuentra disponible en el repositorio Netlib (12).
RESULTADOS Y DISCUSIÓN
Detalles computacionales
Se realizó una serie de cálculos sobre las moléculas H2, HF y H2O empleando una base 6-31G(d,p) para los electrones y una base 1s para los núcleos [6-31G(d,p):1s]; los átomos de flúor (F) y oxígeno (O) se tomaron como cargas puntuales, y los umbrales para la convergencia de la optimización fueron de 1x10-5 y 1x10-6 Hartree/Bohr. Para el cálculo de derivadas numéricas se empleó la fórmula de diferencias finitas centradas (fórmula de los tres puntos). Todos los cálculos fueron realizados en una terminal con procesador Intel(R) Dual Core de 2.8GHz, con sistema operativo GNU/Linux, libre de procesos diferentes a los requeridos por el paquete APMO.
Tiempos de cálculo
La Tabla 1 muestra los tiempos de cálculo y la norma del gradiente al final de la optimización, empleando gradientes numéricos y gradientes analíticos. El umbral de parada para algoritmo de optimización fue de 1x10-5 Hartree/Bohr. En esta tabla, se hace evidente la mejora en el tiempo de cálculo para los sistemas moleculares empleados. En el caso de las moléculas de hidrógeno, el ahorro de tiempo promedio fue de 39%, respecto a un cálculo empleando gradiente numérico. En el caso del agua, el ahorro promedio fue de 20%. Aunque para el agua el ahorro se redujo respecto al conseguido en la molécula de hidrógeno, se espera que en sistemas más grandes la economía en tiempo de cálculo no siga este comportamiento, dado que cuando se aumentan los grados de libertad del sistema, una optimización basada en cálculos de gradiente numérico requiere un número mayor de cálculos de punto sencillo con el fin de calcular las derivadas respecto a posiciones de las funciones base nucleares o de las cargas puntuales.
En la Tabla 1 también se observa que el ahorro conseguido en el tiempo de cálculo se debe exclusivamente a la determinación analítica de las derivadas de las integrales moleculares, dado que el número de iteraciones durante la optimización es el mismo para las dos formas de cálculo del gradiente. Igualmente, dado que la norma del gradiente se encuentra dentro del mismo orden de magnitud en los dos tipos de cálculo (por lo menos para el H2 y el H2O), se puede decir que el método de optimización sólo se ve afectado en la velocidad a la cual puede disponer del gradiente en el siguiente paso de la optimización, y no en las propiedades inherentes al algoritmo de minimización. De la misma tabla, los cálculos que emplean gradiente numérico para el HF muestran mejor respuesta en tiempo para el HF y su isotopólogo con tritio TF.
En la Tabla 2, se muestran los resultados obtenidos para los mismos sistemas, con un umbral de parada para el algoritmo de minimización de 1x10-6 Hartree/Bohr. Los resultados indican que a medida que se refina la búsqueda del mínimo, los cálculos basados en la determinación de gradientes analíticos son superiores a los que emplean gradientes numéricos. Como ejemplo puede observarse que el ahorro en tiempo de cálculo para el H2O pasó de 20% a 65%. De la misma manera se observan mejoras en los cálculos del HF, en los que el DF y el TF encuentran el mínimo con mayor rapidez. En la mayoría de los casos la norma del gradiente al final de la optimización fue más baja para los cálculos que usan gradiente analítico respecto a los que emplean gradiente numérico, lo cual indica que para umbrales de convergencia más bajos, los mínimos encontrados mediante gradientes analíticos son mejores que los obtenidos mediante técnicas numéricas, ya que las fuerzas finales sobre cada átomo son más pequeñas cuando se utiliza el método analítico. La comparación de las Tablas 1 y 2 pone en evidencia la superioridad del empleo del método de gradiente analítico de la energía cuando se desea mejorar la búsqueda de un estado estacionario de un sistema dado.
CONCLUSIONES
Con el fin de buscar una mayor eficiencia en la optimización de geometrías moleculares utilizando el método OMNE, se dedujo e implementó la expresión general del gradiente analítico para cualquier tipo de partícula cuántica. Los cálculos realizados con el fin de comparar la eficiencia del proceso de optimización de las posiciones espaciales de las funciones base nucleares y de los centros de las cargas puntuales, muestran que la determinación analítica del gradiente de la energía presenta mejoras respecto al tiempo de optimización de cálculos que usan métodos numéricos. Adicionalmente, dado que el gradiente analítico de la energía se obtiene mediante la expresión analítica deducida en este trabajo, los estados estacionarios obtenidos son más exactos que los encontrados mediante el método numérico. Los resultados de los sistemas tratados indican que la determinación analítica del gradiente produce mejoras en el tiempo de cálculo, sin afectar el método de minimización multidimensional propiamente dicho.
Con la deducción e implementación de las expresiones analíticas para el cálculo del gradiente de la energía dentro la teoría OMNE/RHF, se ha completado la primera versión del paquete computacional APMO, la cual se pondrá próximamente a disposición de la comunidad académica.
AGRADECIMIENTOS
La presente investigación fue financiada por la División de Investigaciones Sede Bogotá (DIB), de la Universidad Nacional de Colombia, proyectos 20101007499 y 20101009155, a la que los autores expresan su agradecimiento.
REFERENCIAS BIBLIOGRÁFICAS
1. Pulay, P. Ab initio calculation of force constants and equilibrium geometries in polyatomic molecules. Mol. Phys. 1969. 17: 197-204. [ Links ]
2. Yamaguchi, Y.; Goddard, J. D.; Osamura, Y.; Schaefer, H. F. A new dimension to quantum chemistry analytic derivative methods in ab ini-tio molecular electronic structure theory. New York, Oxford University Press. 1994. [ Links ]
3. Nakai, H.; Ikabata, Y.; Tsukamoto, Y.; Imamura, Y.; Miyamoto, K.; Hoshino, M. Isotope effect in dihy-drogen-bond systems: application of analytical energy gradient method in the nuclear orbital plus molecular orbital theory. Mol. Phys. 2007. 105: 2649-2657. [ Links ]
4. Webb, S. P.; Iordanov, T.; Hammes-Schiffer, S. Multiconfigurational nuclear-electronic orbital approach: incorporation of nuclear quantum effects in electronic structure calculations. J. Chem. Phys. 2002. 117: 4106-4118. [ Links ]
5. Tachikawa, M.; Mori, K.; Nakai, H.; Iguchi, K. Anextention of ab initio molecular orbital theory to nuclear motion. I. Chem. Phys. Lett. 1998. 290: 437-442. [ Links ]
6. Tachikawa, M. Multicomponent molecular orbital theory for electron and nuclei including many-body effect. Chem. Phys. Lett. 2002. 360: 494-500. [ Links ]
7. Nakai, H. Simultaneous Determination of nuclear and electronic wave functions without Born-Oppenheimer Approximation: Ab Initio NO + MO/HF Theory. Int. J. Quant. Chem. 2002. 56: 511-517. [ Links ]
8. Nakai, H. Nuclear Orbital Plus Molecular Orbital Theory: Simultaneous Determination of Nuclear and Electronic Wave Functions Without Born-Oppenheimer Approximation. Int. J. Quant. Chem. 2007. 107: 2849-2869. [ Links ]
9. González, S. A.; Aguirre, N. F.; Reyes, A. APMO: un programa computacional para el estudio de efectos cuánticos nucleares mediante la teoría del orbital molecular electrónico y no electrónico. Rev. Colomb. Quim. 2008. 37: 93-103. [ Links ]
10. González, S. A.; Aguirre, N. F.; Reyes, A. Theoretical investigation of isotope effects: The Any-Particle Molecular Orbital code. Int. J. Quant. Chem. 2008. 108: 1742-1749. [ Links ]
11. Obara, S.; Saika, A. Efficient recursive computation of molecular integrals over Cartesian Gaussian functions. J. Chem. Phys. 1987. 84: 3963-3974. [ Links ]
12. Netlib Repository [online]. AT&T Bell Laboratories, University of Tennessee and Oak Ridge National Laboratory. Mayo 2008 [citada 10-05-2008]. Disponible en internet en http://www.netlib.org/tom/500. [ Links ]

