










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Química
versión impresa ISSN 0120-2804versión On-line ISSN 2357-3791
Rev.Colomb.Quim. v.38 n.1 Bogotá ene./abr. 2009
PROTÓN AFFFNITY AND PROTONATION SITES EN P-NITROANILFNE
AFINIDAD PROTÓNICA Y SITIOS DE PROTONACIÓN EN P-NITROANILINA
AFINIDAD PROTÓNICA E LUGARES DE PROTONACIÓN EM P-NITROANILINE
Cinthia Jaramillo, Luis Fernando Moreno, Albeiro Restrepo1
1 Instituto de Química, Universidad de Antioquia, AA 1226, Medellín, Colombia. albeiro@matematicas.udea.edu.co
Recibido: 07/11/08 - Aceptado: 03/04/09ABSTRACT
We present a study of the effect that substituents with competitive inductive properties have on the reactivity of the aromatic ring. As specific case, we studied the gas phase protonation of paranitroaniline (PNA) using Density Functional Theory methods (DFT) and Moller Plesset second order perturbation Theory (MP2). The joint effect of the substituents yields relative stabilities on the protoanted species that follow the trend nitro group > amino group > ortho carbon > para carbon > meta carbon > ipso carbon (positions relative to the amino group).
Key words: Proton affinity, reactivity in aromatic rings, competitive inductive effects, p-Nitroanline.
RESUMEN
Se presenta un estudio del efecto de sustituyentes con propiedades inductivas competitivas sobre la reactividad en el anillo aromático. Como caso específico se estudió la protonación de la para nitro anilina (PNA) en fase gaseosa utilizando métodos de teoría de los funcionales de la densidad (DFT) y teoría de perturbaciones de segundo orden bajo el formalismo de Moller Plesset (MP2). El efecto conjunto de los sustituyentes conduce a estabilidades relativas para las especies protonadas de acuerdo con la siguiente escala: grupo nitro > grupo amino > carbono orto > carbono para > carbono meta > carbono ipso (posiciones relativas al grupo amino).
Palabras clave: afinidad protónica, reactividad de anillos aromáticos, efectos inductivos cooperativos, p-Nitroanilina.
RESUMO
A presenta um estudo do efeito de substituintes com propriedades indutivas competitivas sobre a reação no anel aromático. Como caso específico se estudou a protonação da para nitro anilina (PNA) em fase gasosa utilizando métodos de Teoria dos Funcionais da Densidade (DFT), e Teoria de Perturbações de segunda ordem debaixo o formalismo de Moller Plesset (MP2). O efeito conjunto dos substituintes conduz a estabilidades relativas para as espécies protonadas de acordo com a seguinte escala: grupo nitro > grupo amino > carbono orto > carbono para > carbono meta > carbono ipso (posições relativas ao grupo amino).
Palavras-chave: afinidad protónica, reactividad do anel aromático, efeitos indutivos competitivos, p-Nitroanilina.
INTRODUCTION
In Organic Chemistry, substituent effects are traditionally described in a qualitative fashion. Substituent inductive effects are helpful in predicting the behavior and reactivity of many molecules, but they are not enough for a complete description in the cases where multiple substitution with opposite effects are present. For example, relative basicity criteria establish that aniline protonation is favored on the nitrogen atom of the amino group, predictions that are in agreement with liquid phase experimental results (1). On the other hand, gas phase protonation sites of aniline is still a subject of debate: two protonated isomers have been found experimentally (2); computationally, coexistence of two isomers with slight energy preference towards protonation on the para carbon to the amino group is predicted (3). Regarding paranitroanili-ne, experimental studies suggest that the preferred protonation sites are the oxygens belonging to the nitro group (4,5).
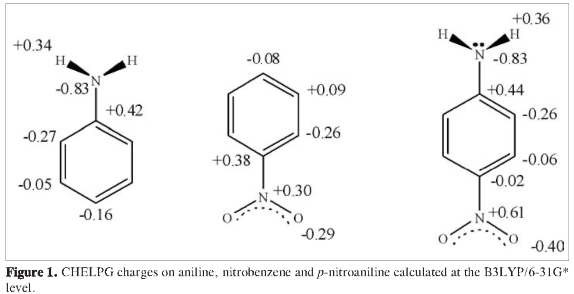
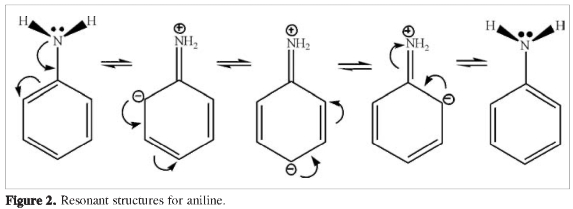
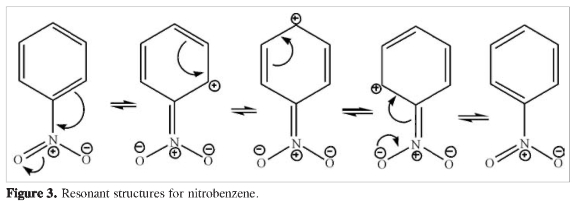
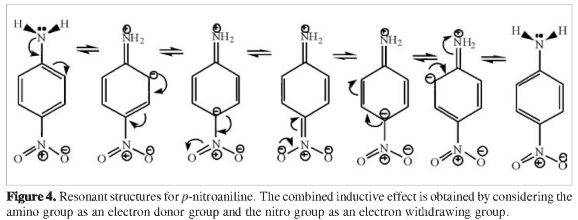
The various inductive effects on aniline, nitrobenzene and paranitroaniline are depicted in Figures 1-4. Atom charges (Figure 1) were calculated under the CHELPG fitting scheme to the electrostatic potential (6), resonant structures for aniline (Figure 2), nitrobenzene (Figure 3) and p-nitroaniline (Figure 4) are drawn considering -NH2 as an electron donor group and -NO2 as an electron withdrawing group. Charge distribution on the ring in paranitroaniline resembles the charge distribution in aniline, which suggests a dominant effect from the amino group. The significant negative charges on the nitrogen and oxygen atoms and on the ortho carbon on p-nitroaniline makes them favorable sites for proton attacks. Resonant structures predict induced negative charges on the aromatic ring on para-nitroaniline that resemble those produced on aniline while the induced charges on nitrobenzene and p-nitroaniline differ markedly; this observation again suggests that the amino group has an inductive dominant effect over the nitro group. The resonance induced reactivity on the ortho position on the ring seems to be in good agreement with the reactivity towards electrofilic attack as predicted by the charge distribution, however, the reactivities on the para position predicted by the two criteria are in conflict: the resonant structures favor proton attack (there are contributing structures bearing negative charges on the para carbon) while the charge analysis predicts no enhanced effect on that position; furthermore, the resonance criterion makes no difference regarding the possibility of proton attack on the ortho and para positions. From the above statements it is observed that using qualitative inductive effects to predict reactivity on the aromatic ring when substituents with opposing effects are present yields conflicting and inconclusive results.
In order to have a complete picture of the preferences for protonation sites, it is necessary to relate not only inductive effects and relative stabilities of the products (thermodynamic criterion) but also the feasibility for each reaction as predicted from rate constants (kinetic criterion). In this work we study at the B3LYP/ 6-31g* and MP2/6-31g* levels the relative stabilities of the reactants and products for all the possible protonation sites in paranitroanline aiming at describing from the thermodynamic point of view the preferences for protonation sites, which can not be established a priori from qualitative inductive effects.
METHODOLOGY
Stationary points within Potential Energy Surfaces (PES) for all the possible protonated p-nitroanlines corresponding to well characterized minina (no negative eigenvalues of the Hessian matrix) were located at the B3LYP (7-9) and MP2 (10, 11) levels in conjunction with the 6-31g* basis set. This choice of methodology has proven successful in matching experimental data for aniline protonation (3, 12). All geometry optimizations and characterization of stationary points via analytical harmonic vibrational frequencies were carried out using the Gaussian03 suite of programs (13). Following standard practice (see for example ref. 14), zero point corrected proton affinities (PA) for a molecule M were calculated as
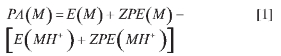
Where E(M) and E(MH+) are the electronic energies for the unprotonated and pro-tonated molecules respectively and the ZPEs are their corresponding corrections for the Zero Point vibrational Energies. Atom charges were calculated under the CHELPG fitting scheme to the electrostatic potential (6). Isomer populations, xi, within the protonated p-nitroanilines PES were estimated by Boltzmann distributions (15-17).
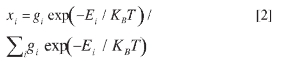
where gi is the symmetry number for isomer i (its degeneracy) and Ei the corresponding B3LYP or MP2 energies.
RESULTS AND DISCUSIÓN
Geometries
MP2 and B3LYP exhibit similar PESs: we located the same minima in both surfaces. There are not significant differences in geometrical parameters between stationary points calculated with either method. In all but one structure, protonated or not, the nitro group is coplanar with the aromatic ring, the one exception being the geometry for the p-nitroaniline protonated in the para positon (relative to -NH2), when at the MP2 level, the nitro group aligns itself with a plane perpendicular to the benzene ring that bisects it from the ipso to the para carbons, while at the B3LYP level, the nitro group retains the orientation it had on the p-nitroanline but elevates from the plane. The aromatic ring suffers little distortion from planarity regardless of the position of incorporation of the proton; if protonation occurs in one of the carbon atoms belonging to the ring, CC bonds on that atom are elongated while the other CC bonds within the ring are somewhat shortened; in the case of ipso protonation, the CN bond (to the -NH2 group) remains almost unchanged when compared to the CN bond in p-nitroaniline, however if protonation occurs in the para position, the CN bond (to the -NO2 group) is enlarged by 0.1 Å (1.46 vs 1.56 A, MP2). The hydrogen atoms belonging to the amino group are deviated from the benzene plane by about 30 degrees in the unprotonated species; in all but two cases, protonated species place those hydrogens in the plane no matter how far from the amino group the protonation happens, the exceptions being protonation on the ipso position and in the amino group itself. Overall, when protonation happens on the ring, the atom or group bonded to the carbon atom that accepts the proton suffers the most displacement from its original position.
Energetics
Tables 1 and 2 summarize the energy analysis for the gas phase protonation of p-nitroaniline at the B3LYP/6-31g* and MP2/6-31g* levels respectively. It is observed that relative stabilities follow the same trend in both cases although energy differences among isomers are not the same. In both PESs, the most stable pro-tonated structure by far is the one that incorporates the proton on the nitro group, this result is in contradiction with the charge analysis, which suggests that protonation should be favored on the nitrogen atom of the amino group (Figure 1). The stability of protonated p-nitroanlines according to the protonation site follows the order -NO2 > -NH2 > ortho > para > meta > ipso. In contrast with B3LYP, MP2 calculations predict the PNA proto-nated at the amino group to be somewhat close in energy to the PNA protonated at the nitro group, (ΔPA = 4.91 at MP2 vs 15.17 at B3LYP); in any case, the energy difference is still too large in favor of the PNA protonated at the nitro group as to render its population close to 100 %. Regarding activation of the ring towards electrophilic attack by considering only the species protonated at the ipso, ortho, meta and para positions, a number of observations can be drawn from Tables 1 and 2: (a) There is no activation of the ipso and meta positions, (b) the ortho position is heavily favored, however, there is some possibility of protonation at the para position, (c) protonation at the ipso and meta positions from thermodynamic criteria alone are disfavored by circa 20 and 37 kcal/mole respectively.
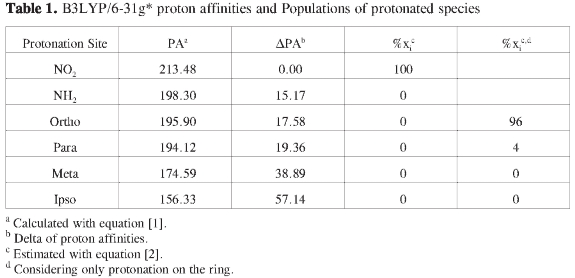
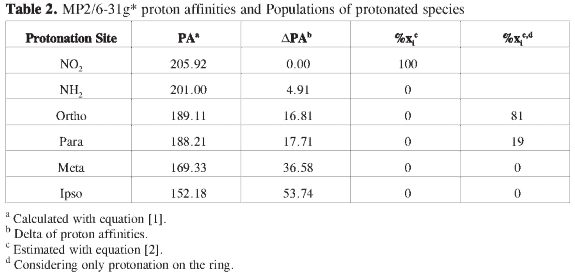
The results on Tables 1 and 2, related to electrophilic attack, could be better rationalized using the inductive effects arising from resonant structures (Figure 4) than using the charge distribution (Figure 1): negative charges are prominent on the amino, nitro groups and on the ortho carbon and negligible at the meta and para carbons, the ipso carbon bears a large positive charge, however, the PNA protonated at the para position is very close in energy to the PNA protonated at the ortho position. Neither the charge distribution nor the resonant structures can account for the presence of well characterized minima on the PES of protonated structures at the ipso and meta positions. It was mentioned on the introduction that both the charge distribution and resonant structures are dominated by the presence of the amino group (Figures 1-4), however, experimental results for aniline favor protonation on the nitrogen atom of the amino group and on the para position, while computational results disfavor protonation at the ortho position in aniline (3); our results indicate a high probability of proton attack at the ortho positions for the PNA, clearly this in contradiction with the suggested dominance of the amino group.
CONCLUSIONS AND PERSPECTIVES
Thermodynamic analysis for the gas phase protonation of p-nitroanline at the B3LYP/6-31g* level gives similar trends as those predicted by the more expensive MP2/6-31g* calculations. Both B3LYP and MP2 methods predict the -NO2 group to be the most favorable site for proton attack on p-nitroaniline; these results are in excellent agreement with expertimental evidence. There is some conflict between reactivity predicted by charge analysis and by resonant structures, the amino group seems to dominate in both pictures, however, neither approach can fully explain the calculated relative stabilities nor the protonation on the ipso and meta positions, predicted here to be feasible processes. Our results are by no means conclusive because a wider view that includes kinetic effects is needed for a proper description of the reactions, the thermodynamic criterion alone only ensures that the products are stable, it does not say anything about the processes abilities to occur, we are currently working to address the kinetic issues.
ACKNOWLEDGEMENTS
Funding for this project by the Comité para el Desarrollo de la Investigación CODI, at the University of Antioquia is acknowledged. We thank professor Jorge Zuluaga and the Physics Institute at (U de A) for ample provisions of computer time at the Hercules and Zipa clusters.
REFERENCES
1. McMurry, J. Organic Chemistry. 4th ed. Pacific Grove, CA.: Brooks/Cale. 1996. [ Links ]
2. Karpas, Z.; Berant, Z.; Stimac, R. An Ion Mobility Spectrometry/Mass Spectrometry (IMS/MS) study of the site of protonation in anilines. Struc. Chem. Soc. 1990. 1: 201-204. [ Links ]
3. Russo, N.; Toscano, M.; Grand, A.; Mineva, T. Proton affinity and protonation sites of aniline. Energetic behavior and density functional reactivity indices. J. Phys. Chem. A. 2000. 104: 4017-4021. [ Links ]
4. (a) Lau, Y.; Nishizawa, K.; Tse, A.; Brown, R.; Kebarle, P. Protonation and site of protonation of anilines. Hydration and site of protonation after hydration. J. Am. Chem. Soc. 1981. 103: 6291-6295. [ Links ]
5. Lau, Y.; Kebarle, P. Substituent effects on the intrinsic basicity of benzene: proton affinities of substituted benzenes. J. Am. Chem. Soc. 1976. 98: 7452-7453. [ Links ]
6. Breneman, C.; Wiberg, K. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comp. Chem. 1990. 11: 361-373. [ Links ]
7. Stephens, P.; Devlin, J.; Chabalowski, C.; Frisch, M. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994. 98: 11623-11627. [ Links ]
8. Lee, C.; Yang, W.; Parr, R. Development of the colle-Salvetti correlation-energy formulainto afunctional of the electron density. Phys. Rev. B. 1988. 37: 785-789. ' [ Links ]
9. Becke, A. Density - Functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993. 98: 5648-5652. [ Links ]
10. Moller, C.; Plesset, M. Note on an approximation treatment for many-electron system. Phys. Rev. 1934. 46: 618-622. [ Links ]
11. Krishnan, R.; Pople, J. Approximate fourth-order perturbation theory of the electron correlation energy. Int. J. Quantum Chem. 1978. 14: 91-100. [ Links ]
12. Wiberg, K. Substituent effects on the acidity of weak acids 4: anilinium Ions Coll. Czech. Chem. Com. 2004. 69: 2183-2192. [ Links ]
13. Frisch, M. et al. Gaussian 03, Revision D.01. Wallingford, CT: Gaussian Inc. 2004. [ Links ]
14. Hilldebrand, C.; Klessinger, Eckert-Maksic, M.; Maksic, Z. Theorical model calculations of the proton affinities of aminoalkanes, aniline, and piridine. J. Phys. Chem. 1996. 100: 9698-9702. [ Links ]
15. Albeiro Restrepo. Influence of p Bonds on the Structure of Organic Molecules. Ph. D. Thesis, University of Connecticut, Storrs, CT, 2005. [ Links ]
16. Pérez, J.; Hadad, C.; Restrepo, A. Structural studies of the water tetramer. Int. J. Quantum Chem. 2008. 108: 1653-1659. [ Links ]
17. Pérez, J.; Flórez, E.; Hadad, C.; Fuentealba, P.; Restrepo, A. Stochastic search of the quantum con-formational space of small lithium and bimetallic lithium-sodium clusters. J. Phys. Chem. A. 2008. 112: 5749-5755. [ Links ]

