










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804On-line version ISSN 2357-3791
Rev.Colomb.Quim. vol.38 no.1 Bogotá Jan./Apr. 2009
ESTUDIO TEÓRICO DEL EFECTO ISOTÓPICO DE HIDRÓGENO EN EL ADUCTO BORANO-CARBONILO
THEORETICAL STUDY OF THE HYDROGEN ISOTOPE EFFECT ON THE BORANE-CARBONILE ADDUCT
ESTUDO TEORICO DO EFEITO ISOTOPICO DE HIDROGENO NO ADUCTO BORANO-CARBONILO
Neftalí Forero1, Sergio A. González1, Andrés Reyes1
1 Departamento de Química, Facultad de Ciencias, Universidad Nacional de Colombia, sede Bogotá, Bogotá, Colombia. areyesv@unal.edu.co
Recibido: 27/11/08 - Aceptado: 13/04/09RESUMEN
Se estudió el efecto de la sustitución isotópica de hidrógeno sobre la geometría, la estructura electrónica y la estabilidad del aducto borano-carbonilo, mediante el método de orbitales moleculares nucleares y electrónicos (OMNE) implementado en el paquete computacional APMO. Se encontró que el aumento de la masa isotópica acorta las distancias de enlace boro-isótopo y carbono-oxígeno, mientras que alarga la distancia boro-carbono. Se determinó además la estabilidad del aducto a partir de las energías de formación y las distancias de enlace B-C. Se encontró que el aumento de la masa isotópica debilita el enlace B-C. Una primera explicación de este fenómeno en términos del concepto de acidez de Lewis predijo resultados contrarios a los encontrados, mientras que un modelo de reactividad basado en las diferencias de las energías de los orbitales LUMO del borano permitió dar cuenta de este efecto.
Palabras clave: efecto isotópico de hidrógeno (EI), aducto borano-carbonilo, complejo donor-aceptor electrónico (DAE), método de orbitales moleculares nucleares y electrónicos (OMNE), any particle molecular orbital (APMO), aproximación de Born-Oppenheimer (ABO).
ABSTRACT
We have investigated the hydrogen isotope effect on the geometry, the electronic structure and the stability of the borane-carbonile adduct, by using the nuclear-electronic molecular orbital method (NEMO) which has been implemented in the APMO software. We have found that an increase of the mass of the hydrogen isotope reduces the boron-hydrogen and carbon-oxygen bond lengths while increasing the boron-carbon distance. In this study, the stability of the adduct has been analyzed in terms of formation energies and B-C bond distances. We have found that the increase of the isotope mass weakens the B-C bond. We tried to give an explaination to this phenomenon based on Lewis acidity concept but it predicted the wrong results. A reactivity model based on the energy differences of borane LUMO orbitais offered a correct explaination to this effect.
Key words: hydrogen isotope effect, borane-carbonile adduct, electron donor-aceptor complex, nuclear-electronic molecular orbital method, any particle molecular orbital (APMO), Born-Oppen-heimer approximation (ABO).
RESUMO
Temos estudado o efeito da substituição de isótopos de hidrogénio sobre a geometria, estrutura electrónica e da estabilidade do aduto Borana-carbonil, através do mêtodo de orbitais moleculares-core versões (OMNE) implementado no pacote computacional APMO. Descobrimos que um aumento da isótopo massa encurta as distâncias de ligação boro-carbono-isotópica de oxigênio e aumenta a distância, enquanto boro-carbono. Foi ainda determinada a estabilidade do aduto de as energias de formação e distâncias de ligação BC. Nós descobrimos que o aumento da massa isotópica minar a relação BC. Uma explicação para este fenômeno em termos de conceito de acidez de Lewis predita resultados contrários aos encontrados enquanto um modelo baseado em diferenças de reatividade nas energias dos orbitais LUMO do Borana autorizados a conta para este efeito.
Palavras-chave: efeito de isótopos de hidrogênio, aduto borana-carbonil, doador-aceitante complexo eletrônico, método de orbitais moleculares-core electrónicos, any particle molecular orbital (APMO), aproximación de Born-Oppenheimer (ABO).
INTRODUCCIÓN
Los complejos ácido-base, también conocidos como complejos donor-aceptor electrónico (DAE) (1), han sido ampliamente estudiados con diversos propósitos, tanto experimental como teóricamente. Hasta la fecha se ha realizado un gran número de investigaciones teóricas con el fin de entender y predecir la estructura y estabilidad de los DAE; entre las metodologías más utilizadas se encuentran el análisis energético de Morokuma (2), el modelo geminal extendido de Roeggen (3), el esquema de partición de orbitales naturales de enlace (4) y el análisis topológico de la función de onda de Bader (5, 6), entre otros.
Los aductos del borano (BH3) son ejemplos muy representativos de los DAE, ya que el boro se comporta en estos como un claro aceptor de electrones (ácido de Lewis). En la literatura se ha reportado una gran cantidad de trabajos teóricos y experimentales en los que se investiga el efecto de los sustituyentes en la acidez de Lewis (acidez de ahora en adelante) del boro. Entre los hechos más notables se ha encontrado que la acidez de los haloboranos aumenta en un orden inverso a la electronegatividad del halógeno así: BBr3 >BCl3 >BF3 (7-8).
Hasta donde se tiene conocimiento, no se han realizado estudios teóricos o experimentales acerca del efecto de isótopo de hidrógeno (EI) sobre la acidez del borano. Un estudio teórico de estos efectos requiere una metodología que describa explícitamente el efecto de la masa nuclear sobre la estructura electrónica. En este sentido, los métodos tradicionales de estructura electrónica basados en la ABO no permiten dar una descripción correcta o directa del cambio de la función de onda electrónica con el cambio de la masa del isótopo. En esta aproximación, el cálculo de la función de onda electrónica es completamente independiente de las masas nucleares. Se propone en este artículo realizar un estudio del EI sobre la estructura y estabilidad del aducto borano-carbonilo (aducto en el resto del artículo) utilizando el método de orbitales moleculares nucleares y electrónicos (OMNE) (9), el cual permite calcular directamente los EI a partir de cálculos similares a los de estructura electrónica convencional.
MÉTODOS
Todos los cálculos de estructura electrónica y núcleo-electrónica se realizaron con el programa APMO, el cual implementa el método OMNE a un nivel de teoría Hartree-Fock (HF) para núcleos y electrones. Los detalles completos del método (9-10) y de su implementación computacional se pueden encontrar en el artículo original (11).
RESULTADOS Y DISCUSIÓN
Se empleó una base electrónica 6-31G(d,p) en todos los cálculos reportados eneste artículo. Seusó unabase 1s para representar cada protón, deuterón y tritón (10). Los núcleos de los átomos distintos al hidrógeno se consideraron como cargas puntuales. La tolerancia del gradiente empleada fue 1x10-4 Har-tree/Bohr.
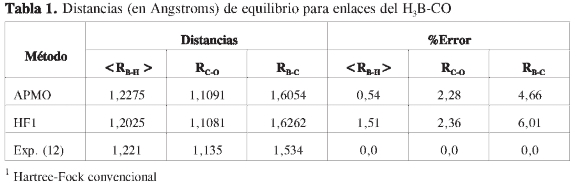
En la Tabla 1 se registran las distancias de enlace del aducto calculadas y experimentales (valores esperados <R> en el caso de núcleos cuánticos), al igual que los errores relativos. La comparación de las distancias del enlace B-C muestra que al nivel de teoría utilizado, la incorporación de los efectos cuánticos nucleares da una mejor correspondencia respecto al valor experimental (error de 0,54%), que el resultado obtenido mediante un cálculo convencional (error de 1,51%); esto indica que la descripción teórica es adecuada para realizar predicciones acerca del EI en la estructura y estabilidad de este aducto. Una comparación entre las distancias calculadas con HF convencional y HF-OMNE hace evidente el efecto de la descripción cuántica de los núcleos sobre la geometría de este tipo de sistemas.
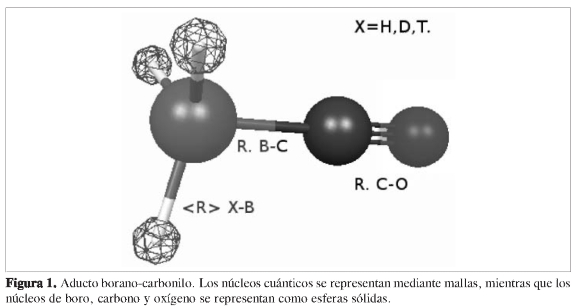
Efecto isotópico sobre la geometría y la estructura electrónica
En la Tabla 2 se presentan las distancias promedio de enlace isótopo-boro, al igual que las cargas de Mulliken sobre el isótopo de hidrógeno y el átomo de boro. Los resultados muestran que mientras aumenta la masa del isótopo, la distancia B-Isótopo disminuye; esta tendencia es la esperada si se considera la naturaleza anarmónica del potencial y la energía de punto cero (ZPE), ya que entre más alto sea la ZPE, mayor será la distancia de enlace.

En la Tabla 2 se observa, además, que a medida que aumenta la masa del isótopo, aumenta la carga negativa sobre este, mientras que se hace menos negativa la carga del boro. Este fenómeno de inducción eléctrica ha sido observado en experimentos de resonancia magnética nuclear sobre moléculas que contienen isótopos de hidrógeno (13), y es una manifestación de los efectos cuánticos nucleares sobre la estructura electrónica.
En la misma tabla se constata que el aumento de masa del isótopo produce una disminución en la distancia de enlace RC-O. Sin embargo, este cambio es en general un orden de magnitud menor al encontrado en la distancia RB-H yRB-C. Este comportamiento es el esperado, ya que los EI secundarios en las distancias de enlace decaen gradualmente con el alejamiento del isótopo. Igualmente se observa en la misma tabla que se presenta un cambio menos drástico de las cargas de Mulliken sobre cada átomo. La disminución progresiva de las cargas con el aumento de la masa del isótopo se interpreta como un efecto netamente inductivo.
Efecto isotópico sobre la estabilidad del aducto
Los EI en la estabilidad del aducto se analizaron en términos de los cambios de la energía de formación y de la distancia de enlace RB-C. Los resultados de la Tabla 3 muestran claramente que el aumento de la masa del isótopo reduce la energía de formación del aducto, lo cual está en concordancia con el incremento gradual de las distancias de enlace RB-C (Tabla 2).
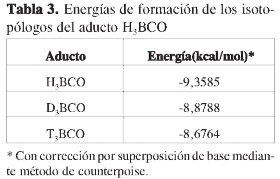
En un primer intento para entender la desestabilización gradual del aducto con el aumento de la masa del isótopo, se recurrió al concepto de acidez de Lewis, según el cual se espera que a menor carga electrónica sobre átomo ácido, mayor será su acidez, y por consiguiente, más fuerte será su enlace con una base de Lewis. Según la Tabla 2, la reducción gradual de las cargas de Mulliken sobre el boro con el aumento de la masa del isótopo sugiere que debería haber un incremento gradual de la acidez del boro. Sin embargo, los resultados de las tendencias de las energías de formación (Tabla 3) y en las distancias de enlace RB-C (Tabla 2) revelan un comportamiento completamente opuesto. Se concluye en este punto que el comportamiento del borano ante una sustitución isotópica no es explicable en términos del concepto de acidez de Lewis. Los resultados de esta investigación indican que existe un debilitamiento del aducto con el aumento de la electronegatividad del isótopo. Un comportamiento similar se ha encontrado en los aductos de haloboranos (X3B-Base, X = F, Cl, Br) (7-8) en los que el aumento en la electronegatividad del halógeno reduce la estabilidad del aducto.
En un segundo intento, se tratará de explicar la desestabilización del aducto con el aumento de la masa del isótopo en términos del estudio de las energías de los orbitales LUMO del borano. De acuerdo con el modelo de reactividad química (14), un LUMO más bajo indica una interacción más fuerte con el orbital HOMO de una especie donora como el carbonilo. En la Tabla 4 se observa que las energías de los LUMO de los boranos aumentan gradualmente con el incremento de las masas de los isótopos, y como consecuencia, según este modelo, debe esperarse una tendencia H3B> D3B> T3B en la fortaleza de los aductos, lo cual está de acuerdo con las tendencias de la energías de estabilización y de las distancias de enlace.

CONCLUSIONES
En este artículo se empleó el método OMNE/HF implementado en el programa APMO para estudiar el efecto isotópico de hidrógeno (EI) en la geometría, la estructura electrónica y la estabilidad del aducto borano-carbonilo.
En un primer estudio se analizó el EI sobre la geometría y la estructura electrónica del aducto. Los resultados mostraron que el aumento gradual de la masa del isótopo acorta las distancias de enlace B-H y C-O, mientras que alarga la distancia B-C. Los EI sobre las cargas de Mulli-ken atómicas hicieron evidente que entre más pesado sea el isótopo, más electronegativo es este.
En un segundo estudio se investigó el EI sobre la estabilidad del aducto. Se encontró que las energías de formación del aducto disminuyen con el aumento de la masa del isótopo. Este resultado esta de acuerdo con la tendencia de la distancia de enlace RB-C. Ambos resultados llevaron a concluir que el aumento de la masa del isótopo desestabiliza el aducto.
Para dar explicación a este fenómeno se acudió al concepto de acidez de Lewis, pero este lleva a predicciones contrarias a las observadas. El concepto de reactividad química, el cual en este caso está dado en términos de las energías de los orbitales LUMO de borano, permitió ver que la disminución el efecto del isótopo sobre los orbitales LUMO sigue las tendencias de energía de estabilización y distancias de enlace encontradas.
Estos resultados permiten concluir que el programa APMO es adecuado para estudiar efectos de isótopo. Los errores observados en las distancias de enlace muestran que para tener una descripción correcta de estos sistemas se requiere incluir correlación electrónica, nuclear y núcleo-electrónica. La implementación del método OMNE a un nivel de teoría MP2 se está realizando actualmente.
AGRADECIMIENTOS
La presente investigación fue financiada por la División de Investigación de la Universidad Nacional de Colombia, sede Bogotá, proyecto 20101009155, a la que los autores expresan su agradecimiento.
REFERENCIAS BIBLIOGRÁFICAS
1. Briegleb, G. Elektronen-Dona-tor-Acceptor-Komplexe. Gottingen: Springer-Verlag. 1961. [ Links ]
2. Umeyama, H.; Morokuma, K. Molecular orbital studies of electron donor-acceptor complexes. 3. Energy and charge decomposition analyses for several strong comple-xes:OC-BH3, H3N-BH3, CH3H2N-BH3, (CH3)3N-BH3, and H3N-BF3. J. Am. Chem. Soc. 1976. 98(23): 7208-7220. [ Links ]
3. Roeggen, I. Derivation of an extended geminal model. J. Chem. Phys. 1983.79(11): 5520-5531. ' [ Links ]
4. Reed, A. E.; Curtiss, L. A.; Wein-hold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988. 88(6): 889-926. [ Links ]
5. Dapprich, S.; Frenking, G. Investigation of donor-acceptor interactions: A charge decomposition analysis using fragment molecular orbitals. J. Phys. Chem. 1995. 99(23): 9352-9362. [ Links ]
6. Bader, R. F. W. Atoms in molecules. Acc. Chem. Res. 1985. 18(1): 9-15. [ Links ]
7. Anane, H.; Boutalib, A.; Tomás, F. Stability of borane-adduct complexes: A G-2 Molecular Orbital study. J. Phys. Chem. A. 1997. 101(42): 7879-7884. [ Links ]
8. Brinck, T.; Murray, S. J.; Politzer, P. A Computational analysis of the bonding in boron trifluoride and boron trichloride and their complex wiht ammonia. Inorg. Chem. 1993. 32(12): 2622-2625. [ Links ]
9. Nakai, H. Simultaneous determination of nuclear and electronic wave functions without Born-Oppenhei-mer approximation: Ab initio NO + MO/HF theory. Int. J. Quant. Chem. 2002. 86(6): 511-517. [ Links ]
10. González, S.; Aguirre, N.; Reyes, A. Theoretical investigation of isotope effects: The Any-Particle Molecular Orbital code. Int. J. Quant. Chem. 2008. 108(10): 1742-1749. [ Links ]
11. González, S. A.; Aguirre, N. F.; Reyes, A. APMO: un programa computacional para el estudio de efectos cuánticos nucleares mediante la teoría del orbital molecular electrónico y no electrónico Rev. Colomb. Quím. 2008. 37(1): 93-103. [ Links ]
12. Rayon, V. M.; Sordo, J. A. On the nature of the interaction in donor-acceptor van der Waals complexes: BH3-CO,BF3-CO,BH3-NH3,B F3-NH3. J. Mol.Struct. (Theo-chem). 1998. 426(1-3): 171-179. [ Links ]
13. Vener, M. V. Modelstudyofthepri-mary H/D isotope effects on the NRM chemical shift in strong hydrogen-bonded systems. Chem. Phys. 1992. 166(3): 311-316. [ Links ]
14. Fukui, K. Theory of Orientation and Stereoselection. Acc. Chem. Res. 1971. 4(2): 57-66. [ Links ]

