










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804On-line version ISSN 2357-3791
Rev.Colomb.Quim. vol.38 no.1 Bogotá Jan./Apr. 2009
ESTUDIO DEL EFECTO DE ISOTÓPO DE HIDRÓGENO EN LOS COMPLEJOS M-HH-F (M=Li, Na)
INVESTIGATION OF THE HYDROGEN ISOTOPE EFFECT ON THE M-HH-F (M=Li, Na) COMPLEXES
ESTUDO DO EFEITO DO ISÓTOPO DO HIDROGÊNIO NO COMPLEXOS M-HH-F (M=Li, Na)
Ismael Ortiz-Verano1, Sergio A. González1, Andrés Reyes1
1 Departamento de Química, Facultad de Ciencias, Universidad Nacional de Colombia, sede Bogotá, Bogotá, Colombia. areyesv@unal.edu.co
Recibido: 27/11/08 - Aceptado: 27/03/09RESUMEN
Se estudió teóricamente el efecto de isotópo de hidrógeno sobre la geometría, la distribución de carga electrónica, la estabilidad relativa y la energía de formación de complejos lineales tipo M-X-Y-F y todos sus isotopólogos de hidrógeno (M=Li, Na; X, Y= H, D, T). Estos estudios fueron realizados con el paquete computacional APMO a un nivel de teoría Hartree-Fock electrónico y nuclear. Los resultados obtenidos son acordes con resultados reportados por otros autores que usan métodos de estructura electrónica convencional.
Palabras clave: efectos cuánticos nucleares, programa APMO, efecto de isótopo, puente de dihidrógeno, aproximación de Born-Oppenheimer.
ABSTRACT
We have carried out a theoretical investigacion of the hydrogen isotope effects on the geometry, the electronic charge distribution, the relative stability and the energy of formation of linear complexes of the type M-X-Y-F and all their hydrogen istopologues (M = Li, Na; X, Y= H, D, T). For this study we have utilized the APMO software at a nucleo-electronic Hartree-Fock level of theory. Our results are agreement with other reported theoretical data based on conventional electronic structure methods.
Key words: nuclear quantum effects, APMO software, isotope effect, dihydrogen bond, Born-Oppenheimer approximation.
RESUMO
Foi estudado teoricamente o efeito do isótopo de hidrogênio sobre geometria, distribuição do carga eletrônica, a estabilidade relativa da energia e da formação do complexos lineares do tipo M-X-Y-F e todos os seus isotopólogos do hidrogênio (M = Li, Na, X, Y = H, D, T). Estes estudos foram realizados com o pacote computacional APMO para um nível da teoria Hartree-Fock eletrônica e nuclear. Os resultados são do acordo com os resultados relatados por outros autores que utilizam métodos do estrutura eletrônica convencionais.
Palavras-chave: efeitos quânticos nucleares, APMO software, efeito do isótopo, puente de di-hidrôgenio, aproximação do Born-Oppenheimer.
INTRODUCCIÓN
El estudio de los puentes de hidrógeno (PH) sigue siendo un área de investigación muy activa y relevante tanto en la química como en la biología (1). La importancia de su estudio radica en que los PH son responsables, entre otros, de la estructura y las propiedades del agua como solvente, y de biomoléculas como el ADN.
Un PH se define generalmente como la interacción atractiva entre dos especies o regiones moleculares en las cuales al menos una de ellas contiene un átomo de hidrógeno (2). Un PH es de la forma D-H-A, en donde la molécula D-H hace las veces de donor, mientras que A hace las veces de aceptor. La mayoría de los PH que se encuentran en la naturaleza involucran átomos de N, O y F (en las posiciones D y A) y son llamados PH convencionales. Sin embargo, existe un gran número de sistemas unidos por PH no convencionales, es decir, aquellos que no pueden ser descritos de la forma D-H-A o aquellos en donde D y A pueden ser átomos de carbono, halógenos, enlaces π, entre otros. Uno de los PH no convencionales que más llama la atención es el llamado puente de dihidrógeno (PDH), M-H-H-Z (M = metal); en este, la molécula Z-H actúa como donor mientras que el hidrógeno de M-H actúa como aceptor.
En este trabajo se centró la atención en el estudio de los efectos de isótopo de hidrógeno en la estructura y estabilidad de los complejos unidos por PDH M-H H-F (M=Li, Na). Dadas las diferencias de electronegatividad entre M y F, se espera que el átomo de hidrógeno unido al átomo alcalino se comporte como el aceptor de puente de hidrógeno, mientras que el perteneciente al fluoruro de hidrógeno se comporte como el donor.
Este tipo de sistemas ha sido estudiado teóricamente por otros autores (3, 4, 5) empleando métodos basados en la aproximación Born-Oppenheimer (ABO). Sin embargo, hasta la fecha, son pocos los estudios teóricos reportados que incluyen efectos isotópicos. Esto se debe en parte a que dentro de la ABO no se tienen en cuenta los efectos cuánticos nucleares sobre la estructura electrónica del sistema molecular, y como consecuencia, cualquier cambio en la estabilidad de los sistemas unidos por PDH sólo es atribuible a los cambios en las energías de punto cero (ZPE) de los sistemas en cuestión.
Una mejor aproximación para dar cuenta de dichos efectos cuánticos nucleares sin recurrir a correcciones posteriores, consiste en tratar los núcleos más ligeros (en este caso, hidrógeno y sus isótopos) como funciones de onda nucleares. Este procedimiento permite tratar teóricamente y desde primeros principios el efecto producido por sustituciones isotópicas de hidrógeno sobre los puentes de dihidrógeno.
MÉTODOS
Todos los cálculos se realizaron con el paquete computacional APMO (6) a un nivel de teoría Hartree-Fock para electrones y núcleos de hidrógeno. En todos los cálculos se empleó una base electrónica 6-31G(d,p). Los núcleos de hidrógeno se representaron mediante una función gausiana 1s para la cual se tomaron los exponentes: 22,3 para 1H (H), 32,8 para 2H (D) y 41,0 para 3H (T) (7). Los núcleos de litio, sodio y flúor se trataron como cargas puntuales. Las estructuras de los iso-topólogos de los monómeros de LiH, NaH y HF y los complejos M-X-Y-F (M=Li, Na; X, Y = H, D, T) se determinaron utilizando el algoritmo de minimización Broyden-Fletcher-Goldfarb-Shanno (8), con un criterio de finalización basado en una tolerancia del gradiente de 10-5 Hartree/Bohr.
RESULTADOS Y DISCUSIÓN
En la Tabla 1 se reportan las distancias de enlace, las cargas atómicas de Mulliken, los momentos dipolares y las energías para cada uno de los monómeros. Las distancias de enlace se calcularon como el valor esperado de R (<R>) después de optimizar la posición de los centros de las funciones nucleares. Los momentos dipolares se calcularon teniendo en cuenta las densidades electrónicas y nucleares.

Efectos isotópicos en la geometría
En la Tabla 1 se observa que las distancias de enlace de los monómeros disminuyen al aumentar las masas del isótopo de hidrógeno. Al contrastar los resultados de las Tablas 1 y 2, se encuentra en todos los casos que la distancia de enlace Y-F aumenta con la formación del complejo mientras que la distancia M-X disminuye. En la Tabla 2 se aprecia que el aumento de la masa del isótopo donor Y produce aumentos en las distancias X-Y (Figura 1 a y b) y X-F (Figura 1 c y d), mientras que el aumento en la masa del isótopo aceptor X produce una disminución en la distancias X-Y (Figura 1 a y b) y X-F (Figura 1 c y d).


En literatura se reporta un estudio teórico al nivel MP2/6-31G(d) (9), en el cual se observa también que la distancia internuclear Li-X (solamente con X = H) disminuye ligeramente (0,8%) al formarse el complejo, mientras que la distancia Y-F (con Y = H) presenta un aumento de 1,7%. Las distancias internucleares calculadas por los autores concuerdan con las reportadas por Remko, así como con las reportadas por Rozas al nivel HF/6-31G** (10).
Efecto de isótopo en la distribución de cargas
En la Tabla 1 se observa que las cargas de los átomos unidos al isótopo de hidrógeno se hacen más positivas a medida que aumenta la masa de este. Tal comportamiento es una clara manifestación de los efectos cuánticos nucleares sobre la estructura electrónica, que indica que entre más pesado el isótopo, mayor es su electronegatividad.
Al estudiar el efecto del aumento de la masa del isótopo en los momentos dipolares, se encuentra que las tendencias son opuestas para las especies donoras M-X y aceptoras Y-F. Si se considera por un momento que se pueden calcular los momentos dipolares como un simple producto de carga de Mulliken por la distancia de separación entre las cargas, se verá que en el caso del sistema Y-F se produce una disminución gradual del momento bipolar; en este caso, tanto las cargas como las distancias de separación se hacen menores, y como consecuencia, el valor del momento dipolar disminuye.
En el caso de M-X se encontró que el aumento de la masa del isótopo produjo un acortamiento en la distancia, y simultáneamente un aumento en la separación de carga. Sin embargo, se observó que a pesar de ser estos efectos contrarios, se produjo un ligero aumento en el momento dipolar molecular, lo cual indica que el efecto de la masa en la estructura electrónica es más importante que aquel en la distancia.
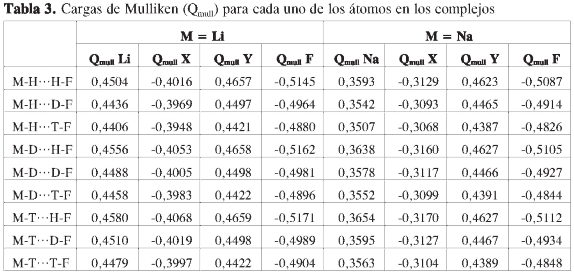
En la Tabla 3 se puede analizar el efecto de la sustitución isotópica sobre las cargas de Mulliken de los átomos en los complejos. Se observa que tanto el aumento de la masa del isótopo donor como la del aceptor produce una disminución en la carga electrónica de los dos átomos unidos a cada uno de estos (efecto primario), y un aumento de la carga electrónica sobre el átomo en posición secundaria. Si se asume que el puente de hidrógeno es de naturaleza dipolar, se puede observar que el aumento de la masa del donor produce una disminución en la separación de cargas, que puede conllevar a la reducción de la interacción dipolar entre los fragmentos moleculares, y de manera contraria, que el aumento de masa del aceptor produce una mayor separación de cargas, y por tanto lleva al fortalecimiento del PDH.
Estabilidad relativa de los isotopómeros de M-X---Y-F (M=Li, Na; X,Y=H,D,TyX≠Y)
Las estabilidades relativas de los isotopómeros M-H-D-F y M-D-H-F, M-H---T-F y M-T---H-F, y M-D---T-F y M-T---D-F se determinaron a partir de la comparación de sus energías calculadas con el paquete computacional APMO (6). Según los datos de la Tabla 2, se observa que los isotopómeros en los que el isótopo más pesado hace las veces de aceptor son más estables que aquellos en los que el isótopo más pesado hace las veces de donor.
Efecto de isótopo en la energía de formación de los puentes de dihidrógeno
En la Tabla 4 se presentan las energías de formación del complejo sin corregir y corregidas del error de superposición de bases electrónicas (11). Se observa claramente que el aumento en la masa del donor debilita el enlace, mientras que el incremento de la masa del aceptor lo fortalece. Estos resultados concuerdan con todos los otros resultados del efecto isotópico en las distancias de enlace, cargas, dipolos y estabilidades relativas. Este tipo de comportamiento también ha sido determinado teóricamente (3,4,5,9)paraotrotipode sistemas unidos por PDH.

CONCLUSIONES
El estudio de estos complejos Li-H-H-F, Na-H-H-F y sus isotopólogos de hidrógeno ha permitido concluir que el aumento de la masa del isótopo donor aumenta la distancia de enlace mientras que disminuye la separación de cargas, el dipolo del complejo y la energía de formación. Estos efectos son totalmente opuestos a los observados al aumentar la masa del aceptor. El presente estudio ha permitido establecer que en el caso de complejos formados con dos isótopos diferentes, habrá una preferencia energética por aquel en el cual el isótopo más pesado hace las veces de aceptor. Estos resultados concuerdan con otros reportados en la literatura y con observaciones experimentales para sistemas similares.
Los resultados presentados en este artículo demuestran el gran potencial del programa APMO para el cálculo de efectos isotópicos de hidrógeno en sistemas unidos por puentes de dihidrógeno. Para una mejor descripción de este tipo de fenómenos se requiere la inclusión de la correlación electrónica, núcleo-electrónica y nuclear. Para lograrla, actualmente se está realizando en nuestro grupo la imple-mentación del método MP2 núcleo-electrónico al paquete computacional APMO.
AGRADECIMIENTOS
La presente investigación fue financiada por la División de Investigaciones de la Universidad Nacional de Colombia, sede Bogotá, proyecto 201010010515, y por COLCIENCIAS, proyecto 1101-40520187, a los que los autores expresan su agradecimiento.
REFERENCIAS BIBLIOGRÁFICAS
1. Scheiner, S. Calculation of isotope effects from first principles. Biochimica et Biophysica Acta. 2000. 1458: 28-42. [ Links ]
2. Kollman, P. A.; Allen, L. C. The theory of hydrogen bond. Chem. Rev. 1972. 72: 285-303. [ Links ]
3. Alkorta, I.; Rozas, I.; Elguero, J. Non-conventional hydrogen bonds. Chem. Soc. Rev. 1998.27: 163-170. [ Links ]
4. Liu, Q.; Hoffman, R. Theoretical aspects of a novel mode of hydrogen hydrogen bonding. J. Am. Chem. Soc. 1995. 117: 10108-10112. [ Links ]
5. McDowell, S. A. C.; Forde, T. S. Isotope effects in linear dihydrogen bonded complexes containing LiH. J. Chem. Phys. 2002. 117: 6032-6037. [ Links ]
6. González, S. A.; Aguirre, N. F.; Reyes, A. Theoretical investigation of isotope effects: The any-particle molecular orbital code. Int. J. Quantum. Chem. 2008. 108: 1742-1749. [ Links ]
7. Ishida, M.; Tachikawa, M.; Tokiwa, H.; Mori, K.; Ishii, A. First principles calculation for hydrogen/positronium os a Si(111) surface using the dynamical extended molecular orbital method. Surf. Sci. 1999. 438: 47-57. [ Links ]
8. Netlib Repository (online). AT&T Bell Laboratories, University of Tennessee and Oak Ridge National Laboratory. Mayo 2008 (citada 10-05-2008). Disponible en internet en http://www.netlib.org/tom/500. [ Links ]
9. Remko, M. Thermodynamics of dihydrogen bonds A-H-H-B. Mol. Phys. 1998. 94: 839-842. [ Links ]
10. Rozas, I.; Alkorta, I.; Elguero, J. Field effects on dihydrogen bonded systems. Chem. Phys. Lett. 1997. 275: 423-428. [ Links ]
11. Boys, S. F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol.Phys. 1970. 19: 553-566. [ Links ]

