










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Química
versión impresa ISSN 0120-2804versión On-line ISSN 2357-3791
Rev.Colomb.Quim. v.38 n.1 Bogotá ene./abr. 2009
EFECTOS COOPERATIVOS EN HETEROTETRÁMEROS (ETANOL)3-AGUA
COOPERATIVE EFFECTS IN (ETHANOL)3-WATER HETEROTETRAMERS
EFEITOS COOPERATIVOS EM HETEROTETRÁMEROS (ETANOL)3-ÁGUA
Sol M. Mejía1, Juan F. Espinal1, Fanor Mondragón1
1 Instituto de Química, Universidad de Antioquia, A.A. 1226, Medellín, Colombia. fmondra@catios.udea.edu.co
Recibido: 07/11/08 - Aceptado: 11/03/09
RESUMEN
La teoría de funcionales de la densidad (DFT: B3LYP/6-31+G(d)) fue empleada para la optimización de agregados estables sobre la superficie de energía potencial de los heterotetrámeros (etanol)3-agua. Las energías de tetramerización pueden llegar a valores hasta de -21,00 kcal/mol. Esta energía no se puede obtener considerando solo contribuciones de interacciones entre dos moléculas del agregado, lo cual sugiere la presencia de efectos cooperativos globales (positivos). Tales efectos son reflejados en distancias menores de los puentes de hidrógeno y distancias menores oxígeno-oxígeno, lo mismo que en elongaciones mayores del enlace O-Hen la molécula dadora de protón con un corrimiento hacia el rojo mayor en los heterotetrámeros, comparado con los heterodímeros de etanol-agua y el dímero de etanol. La mayor cooperatividad fue observada en los cuatro puentes de hidrógeno dispuestos en el patrón geométrico cíclico más grande posible, actuando todas las moléculas como aceptoras y dadoras de protón simultáneamente. Un análisis similar al de la caracterización de heterotetrámeros de (etanol)3-agua se llevó a cabo para los heterotetrámeros (metanol)3-agua y tetrámeros de etanol y metanol. La comparación de estos valores mostró que existe una gran similitud entre todos los parámetros analizados para agregados con el mismo patrón geométrico.
Palabras clave: puentes de hidrógeno, efectos cooperativos, azeótropo etanol-agua, sistemas aceptores-dadores de protón, modelación molecular.
ABSTRACT
Density Functional Theory (DFT: B3LYP/6-31 + G(d)) was used for the optimization of clusters on the potential energy surface of (ethanol)3-water heterotetramers. The tetramerization energies can reach values up to -21.00 kcal/ mol. This energy can not be obtained by just considering the contributions from interactions between two cluster molecules, which suggests of the presence of global cooperative effects (positive). These effects are reflected in smaller hydrogen bond distances and smaller oxygen-oxygen distances, as well as in greater elongations of the O-H proton donor bond with a stronger "red-shift" in the heterotetramers compared to the ethanol-water heterodimers and the ethanol dimer. The largest cooperativity effect was observed in the four hydrogen bonds arranged in the largest possible cyclic geometric pattern, where all the molecules act as proton acceptor and donor simultaneously. A similar analysis to the characterization of (ethanol)3-water heterotetramers was carried out on (methanol)3-water heterotetramers, and ethanol and methanol tetramers, whose comparison showed a great similarity between all evaluated parameters for the clusters with equal geometric pattern.
Key words: hydrogen bonds, cooperative effects, ethanol-water azeotrope, proton donor-acceptor systems, molecular modeling.
RESUMO
A teoria de funcionais de densidade (DFT: B3LYP/6-31+G(d)) foi empregada para a otimização de agregados sobre a superfície de energia potencial dos heterotetrâmeros (etanol)3-água. As energias de tetramerização podem alcançar valores de até -21.00 kcal/mol. Esta energia não pode ser obtida por apenas considerando as contribuições das interações entre agregados de duas moléculas, o que sugere a presença global dos cooperativos efeitos (positivos). Tais efeitos são refletidos em menores comprimentos das pontes de hidrogênio e distâncias oxigênio-oxigênio, e também em maiores alongamentos da ligação O-H na molécula doadora de prótons, com um maior "red-shift" associado nos heterotetrâmeros do que nos heterodímeros de etanol-água e no dímero de etanol. A mais alta cooperatividade foi observada com as quatro pontes de hidrogênio dispostas no maior padrão geométrico cíclico possível, atuando simultaneamente todas as moléculas como aceptoras e doadoras de prótons. Uma análise similar ao da caracterização de heterotetrâmeros de (eta-nol)3-água se levou a cabo sobre heterotetrâmeros de (metanol)3-água e metanol, cuja comparação mostrou uma grande similaridade entre todos os parâmetros analisados para agregados com igual padrão geométrico.
Palavras-chave: ligações hidrogênio, efeitos cooperativos, azeótropo etanol-água, sistemas doador-aceptor de prótons, modelagem molecular.
INTRODUCCIÓN
Los puentes hidrógeno se consideran, en general, las interacciones débiles direccionales más importantes en muchos campos de la ciencia, como la química orgánica e inorgánica, la biología, la química y la física del estado sólido, entre otras (1-3). Por esta razón han sido el centro de una intensa investigación (4-6). Estas interacciones son fundamentales en el estudio de fenómenos tales como la solvatación (7, 8). Por ejemplo, la mezcla azeotrópica etanol-agua presenta una estabilidad inusual frente al calentamien-
to impidiendo su separación por destilación (9, 10). Sin embargo, a pesar de la importancia de los puentes de hidrógeno, muchas características y singularidades de estos sistemas aún no son claras, y en algunos casos son motivo de gran controversia. Algunas de estas particularidades son el inusual corrimiento hacia el azul (11, 12) en algunos puentes de hidrógeno, por ejemplo en sistemas del tipo C-HO, C-H-- π,C-HF y la definición de los criterios para clasificar una interacción como un puente de hidrógeno (4, 13). Incluso, todavía en nuestros días no se ha logrado un consenso para la definición de puentes de hidrógeno, los cuales comúnmente se habían considerado como interacciones con distancias (H---átomo en la molécula aceptora de protón) menores a la suma de los radios de van der Waals. Sin embargo, esta definición ha venido desapareciendo gradualmente de la literatura científica debido a que no cuenta con un buen soporte experimental (14).
En el caso específico de la interacción etanol-agua, de nuestro conocimiento, experimentalmente solo se han estudiado heterodímeros (15, 16). Computacionalmente, el panorama no es mucho más amplio (17-19). Recientemente, se ha reportado que los puentes de hidrógeno del tipo O-H---O, llamados puentes de hidrógeno primarios (convencionales), son los responsables de la estabilidad de los heterotrímeros de (etanol)2-agua (20), mientras que los puentes de hidrógeno del tipo C-H---O, llamados puentes de hidrógeno secundarios (no convencionales (21)), son mucho más débiles que los puentes de hidrógeno primarios, según el análisis de parámetros geométricos y termodinámicos, lo cual también ha sido observado en heterodímeros etanol-agua (17) y en otros sistemas que tienen la posibilidad de formar puentes de hidrógeno no convencionales (22). Como un resultado inherente a la escasa investigación respecto al sistema etanol-agua, los efectos cooperativos no han sido evaluados en este sistema, aunque cuentan con gran reconocimiento en sistemas enlazados por puentes de hidrógeno, tales como agregados de agua (23,24), etanol (25), metanol (26), terbutanol (27) y algunos agregados de metanol/agua (28), ya que a estos se les asigna el reforzamiento o debilitamiento de sus puentes de hidrógeno.
Generalmente, el reforzamiento inusual de estos agregados enlazados por puentes de hidrógeno a medida que crecen en tamaño, se correlaciona con un aumento en la energía de interacción de los mismos (29), los cuales solo pueden ser obtenidos si se considera además de las interacciones entre las moléculas, una expansión mayor de la energía de interacción. Los efectos cooperativos han sido estudiados por medio de la descomposición de la energía de interacción en componentes con significado físico, tales como el método del análisis de la descomposición de energía natural, que ha sido extendido al tratamiento de la densidad de carga por cálculos DFT (30). Este análisis fue el resultado de la búsqueda de otros esquemas de descomposición alternativos al método de Umeyama y Morokuma (31), para el cual ha sido reconocido que puede llevar al tratamiento de una función de onda que viola el principio de exclusión de Pauli (30). Otro modelo es el de muchos-cuerpos TCPE desarrollado por Masella y Flament (27) sobre bases empíricas, que fue aplicado no solo a agregados de agua sino también a agregados de alcohol y heteroagregados alcohol/agua. Los resultados de ambos modelos coinciden en que la estabilización cooperativa en agregados de agua se debe principalmente a la contribución de la transferencia de carga, y en menor grado a la contribución de la polarización inducida de las moléculas. Esto también ha sido observado recientemente por medio de la distribución de cargas de Mulliken en tetrámeros de agua (32), cuyo análisis sugierequelatransferencia de carga es determinante para la estabilización de este sistema.
En este estudio, como un primer acercamiento al entendimiento de los efectos cooperativos en agregados etanol-agua, se caracterizó por medio de modelación molecular heterotetrámeros (etanol)3-agua. La descripción de los efectos cooperativos se realizó sin recurrir a esquemas para la división de la energía. Por tanto, se tuvo en cuenta la explicación cualitativa en términos de argumentos ácido/base propuesta por Mó et ál. (33), la cual es una aproximación que ha proporcionado buenos resultados no solo en trímeros de agua sino también en trímeros de metanol y heterotrímeros metanol/agua (19). La explicación de estos efectos cooperativos también se analizó en términos de la polarización mutua entre las moléculas que forman el agregado, como fue resaltado por Steiner en su revisión acerca de los puentes de hidrógeno en estado sólido (14). Además, para efectos de comparación, se estudiaron hetero-tetrámeros de (metanol)3-agua y tetrámeros de etanol y metanol.DETALLES COMPUTACIONALES
Las geometrías de los agregados fueron totalmente optimizadas con la base 6-31 + G(d) y el funcional B3LYP de la teoría de funcionales de la densidad. Se escogió esta aproximación basados en estudios previos acerca de agregados de (etanol)n-agua (n = 1,2) (20, 34) y en la literatura científica donde se han reportado buenos resultados con el funcional B3LYP (35-37) para describir sistemas enlazados a través de puentes de hidrógeno. Las energías de tetramerización fueron calculadas de la siguiente forma: ΔEtet= Eagregado - ΣEmonomeros; dichas energías incluyen la corrección del error por superposición de las bases (BSSE), usando el método de counterpoise (38). También se realizó la corrección por la energía del punto cero (ZPE), obtenida de los cálculos de frecuencias vibracionales, las cuales fueron para todos los casos frecuencias reales. Por tanto, todas las estructuras reportadas en este estudio corresponden a mínimos sobre sus respec- tivas superficies de energía potencial.
Considerando que Kar y Scheiner (39) encontraron que en los puentes de hidrógeno C-H---O se presentan menos efectos cooperativos que cuando hay puentes de hidrógeno del tipo O-H---O, en este estudio se evaluaron los efectos cooperativos solo para los puentes de hidrógeno primarios. Este objetivo se llevó a cabo por medio del análisis de los cambios en las distancias H-O y O-O, el corrimiento al rojo del modo de estiramiento del enlace O-H en las moléculas dadoras de protón, al igual que su elongación para cada puente de hidrógeno. También, se calculó el factor de cooperatividad Ab,definido como Ab = (ΔvOH)/(Δv'OH) (40, 41), donde se relaciona el corrimiento al rojo de la frecuencia de estiramiento del enlace O-H de la molécula dadora de protón en el heterotetrámero o tetrámero (ΔvOH) con el correspondiente corrimiento al rojo en el heterodímero o dímero (Δv'OH), como un indicativo de la influencia de un primer puente de hidrógeno sobre la formación de un segundo puente de hidrógeno. Todos los cálculos se llevaron a cabo empleando el paquete de programas Gaussian 03. Los órdenes de enlace de los puentes de hidrógeno, tanto primarios como secundarios, se calcularon con el programa NBO.
RESULTADOS Y DISCUSIÓN Energía de tetramerización
El proceso de obtención de estructuras correspondientes a mínimos sobre la superficie de energía potencial de los heterotetrámeros de (etanol)3-agua, se llevó a cabo de forma analítica. Fueron planteados varios posibles arreglos geométricos de las cuatro moléculas, considerando el carácter dador y aceptor de protón debido a los enlaces O-H del agua así como de la molécula trans de etanol. Dicho confórmero de etanol es uno de los mayoritarios junto con los confórmeros gauche (g1, g2) (42-44) y es el más estable, con una diferencia de 41,2 ± 5cm-1 (0,119 ± 0,014 kcal/mol) (45).
Se optimizaron satisfactoriamente cinco heterotetrámeros diferentes con energías de tetramerización entre -20,38 kcal/mol y -13,34 kcal/mol, cuyas geometrías optimizadas se muestran en la Figura 1. Considerando este parámetro termodinámico, las estructuras fueron clasificadas como (a) > (b) ~ (c) ~ (d) > (e) según el orden decreciente en exotermicidad.
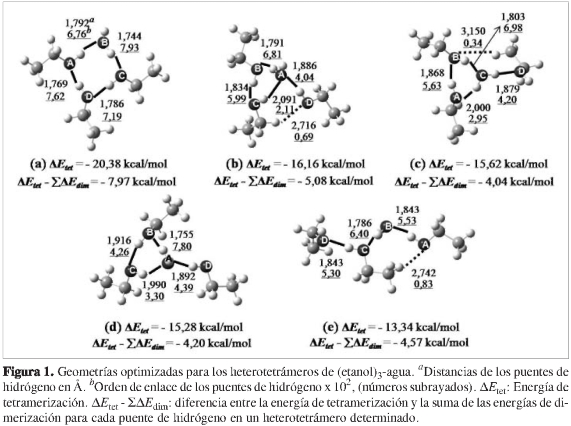
Una vez clasificadas las estructuras de los heterotetrámeros de (etanol)3-agua, se procedió a optimizar las estructuras de los demás tipos de agregados, las cuales fueron nombradas como Et(x), Met(x) y Met-ag(x) para los tetrámeros de etanol y metanol y los heterotetrámeros de (metanol)3-agua, respectivamente, donde la x representa (a), (b), (c), (d) o (e) según estos agregados presenten un patrón geométrico similar a uno de los patrones geométricos encontrados para los hetero-tetrámeros de (etanol)3-agua. Como se puede observar en las Figuras 2, 3 y 4, sólo se lograron optimizar tres tetrámeros de etanol, cuatro tetrámeros de metanol y tres heterotetrámeros de (metanol)3-agua; los otros agregados aparentemente no correspondían a mínimos sobre la superficie de energía potencial. Las energías de tetramerización de estos agregados (ver Figuras 2, 3 y 4) muestran gran similitud con las obtenidas para los heterotetráme-ros de (etanol)3-agua (ver Figura 1), de modo que se conserva la clasificación según el orden decreciente en exotermicidad. Una comparación con los rangos hallados experimentalmente reportados en la literatura científica de las entalpías de tetramerización para el etanol (-19,15 a -25,15 kcal/mol) (46, 47) y el metanol (-20,01 a -26,01 kcal/mol) (6,48,49) muestra que las energías de tetramerización de los agregados más estables para cada caso están entre dichos rangos (Et(a), Met(a)), lo cual es coherente puesto que al ser los agregados más favorables termodinámicamente, se espera que sean los más abundantes.
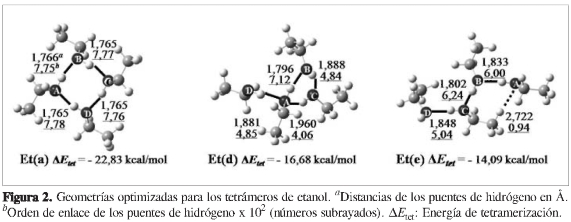
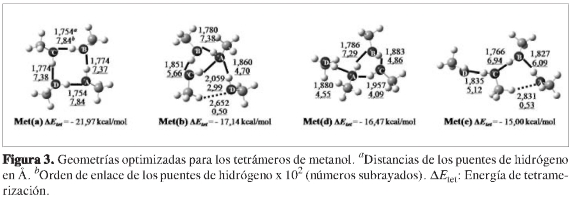
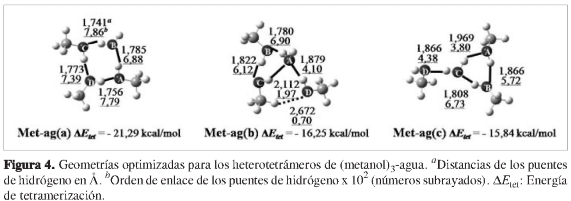
Para todos los casos, se puede asociar una mayor estabilidad con el mayor número de puentes de hidrógeno primarios (líneas negras continuas), dispuestos en el patrón cíclico más grande (estructuras con patrón geométrico (a)), con todas las moléculas actuando como aceptoras y dadoras de protón simultáneamente. Una posible explicación de esta estabilidad se presenta más adelante en la sección de efectos cooperativos. Para este tamaño de agregados, los puentes de hidrógeno secundarios no muestran una contribución a su estabilidad en un grado apreciable; este efecto es similar al encontrado para los heterotrímeros (etanol)2-agua (20) en un estudio previo. Al comparar las dos estructuras con el patrón geométrico (c), (ver Figuras 1 y 4), el heterotetrámero de (metanol)3-agua (ΔEtet = -15,84 kcal/ mol) sin puentes de hidrógeno secundarios es incluso un poco más exotérmico que el heterotetrámero de (etanol)3-agua (ΔEtet = -15,62 kcal/mol), en el cual se formó un puente de hidrógeno secundario entre las moléculas de etanol B y D.
Se puede observar que la energía de tetramerización no puede ser considerada como la suma de las energías de dimerización de los diferentes heterodímeros o dímeros que están presentes dentro de los heterotetrámeros (etanol)3-agua (ver Figura 1 y Tabla 1), ya que esta sumatoria para los cinco heterotetrámeros es menos exotérmica que las respectivas energías de tetramerización, como se muestra en la Figura 1 (ΔEtet - ΣΔAEdim). Valores similares de esta diferencia se pueden obtener para los demás tipos de agregados, por ejemplo para los agregados Et(a), Met(a) y Met-ag(a); la diferencia ΔEtet -ΣΔEdim es de -8,4 kcal/mol, -8,0 kcal/ mol y -7,9 kcal/mol, respectivamente. Por tanto, el análisis de la energía de interacción sirve como indicativo de la presencia de efectos cooperativos globales, en este caso positivos. Ningún agregado presenta efectos cooperativos globales negativos, los cuales son llamados por algunos autores (5, 14) efectos anticooperativos o competitivos.
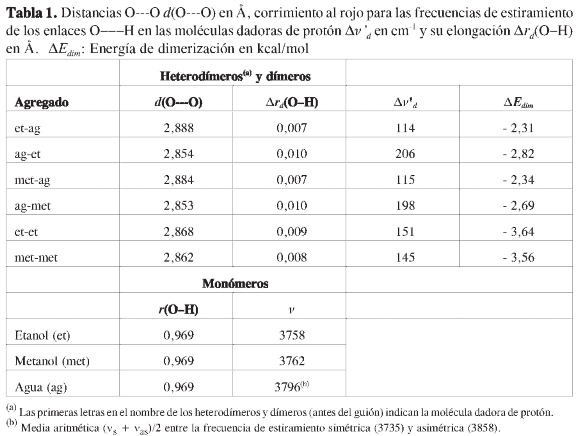
Parámetros geométricos
Los puentes de hidrógeno primarios (representados por las líneas continuas en las Figuras 1-4) más cortos y con mayores órdenes de enlace los presentan en general las estructuras con patrón geométrico (a) (ver Figuras 1 a 4), lo cual puede ser explicado en parte por la linealidad que les permite adoptar el patrón geométrico de un ciclo de cuatro puentes de hidrógeno primarios en comparación con los puentes de hidrógeno doblados en los patrones geométricos de un ciclo de tres puentes de hidrógeno primarios (b), (c) y (d). Sin embargo, al igual que en los heterotrímeros (etanol)2-agua (20), no siempre los puentes de hidrógeno más lineales son más fuertes que los que forman un ángulo menor de 180 grados, como resulta de la comparación de los patrones geométricos (b), (c) y (d) con el patrón geométrico (e).
Como era de esperarse, según el efecto inductivo de electrones de la parte alquílica del etanol, los puentes de hidrógeno entre moléculas de etanol y agua se ven favorecidos cuando la molécula de etanol actúa como molécula aceptora de protón. Una excepción a este hecho es observada en las estructuras de patrón geométrico (c) (ver Figuras 1 y 4) probablemente debido a los efectos cooperativos, los cuales son descritos y evaluados en la siguiente sección.
Por otra parte, si comparan los puentes de hidrógeno primarios más fuertes reportados en trabajos previos acerca de heterodímeros (34) y heterotrímeros (20, 34), en ningún caso se obtienen distancias de puentes de hidrógeno tan pequeñas como 1,7 Å y órdenes de enlace del orden de 7,90 x 10-2, como es el caso de algunos puentes de hidrógeno obtenidos en el presente estudio. Lo mismo se puede concluir si se comparan los puentes de hidrógeno secundarios (representados por las líneas punteadas en las Figuras 1-4). Por consiguiente, la fuerza de los puentes de hidrógeno para la interacción etanol-agua se incrementa con el aumento en el tamaño de los agregados.
No obstante, se pueden observar algunas anomalías en el comportamiento esperado de los puentes de hidrógeno en los patrones cíclicos de tres puentes de hidrógeno en los patrones geométricos (b), (c) y (d), ya que, de acuerdo con los resultados reportados para heterotríme-ros (20), las diferencias pequeñas en la fortaleza de estos tres puentes de hidrógeno primarios podían ser explicadas considerando solo el efecto inductivo de electrones de la parte alquílica de las moléculas de etanol y metanol.
Es interesante observar que al analizar la longitud y el orden de enlace de los puentes de hidrógeno C -H---A (ver Figuras 1 y 4) en el heterotetrámero (etanol)3-agua (b) (2,091 Å, 2,11 x 10-2) y el heterotetrámero (metanol)3-agua (b) (2,112 Å, 1,97 x 10-2), sus fortalezas son comparables con las fortalezas de los puentes de hidrógeno primarios más débiles reportados en los heterotrímeros (20).
De este modo, se sugiere que el análisis geométrico de las estructuras también sirve para determinar la existencia de efectos cooperativos (a nivel local), ya sean positivos o negativos, los cuales parecen ser cada vez más significativos con el aumento en el tamaño del agregado, independientemente del tipo de moléculas de alcohol de las que esté compuesta dicha estructura, teniendo en cuenta los resultados de Masella y Flament (27) para agregados cíclicos de metanol, etanol y terbutanol de 2 a 6 moléculas con el modelo TCPE.
Efectos cooperativos
Considerando la gran similitud observada entre todos los parámetros analizados en las dos secciones anteriores (termodinámicos y geométricos) para cada patrón geométrico, sin importar el tipo de agregado, en esta sección solo se evalúan los cuatro índices que se muestran en la Tabla 2 para los cinco heterotetrámeros (etanol)3-agua y los tetrámeros de etanol y metanol, al igual que el heterotetrámero (metanol)3-agua con el patrón geométrico (a).
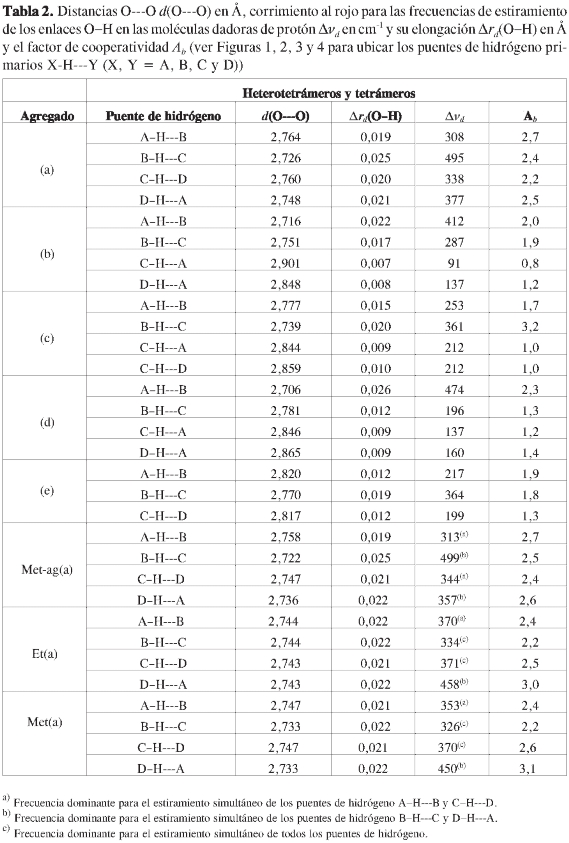
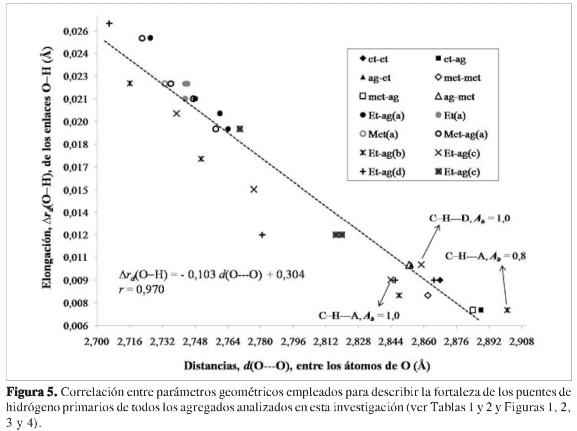
La cooperatividad en estos sistemas se observa en las variaciones en las geometrías de las moléculas de etanol, metanol y agua, las cuales son causadas por la formación de múltiples puentes de hidrógeno (ver Tabla 2). Uno de los cambios observados es en general una mayor elongación del enlace O-H en la molécula dadora de protón en heterotetrámeros y tetrámeros en comparación de heterodímeros y dímeros (ver Tablas 1 y 2), lo mismo que una menor distancia oxígeno-oxígeno. Una comparación entre estos dos parámetros geométricos es graficada en la Figura 5. Se observa que hay efectivamente una correlación lineal inversamente proporcional (Δrd(O-H) = -0,103 d(O---O) + 0,304, r = 0,970) entre estos dos parámetros al graficar sus valores para los puentes de hidrógeno primarios, tanto de los agregados con etanol como con metanol. Esta correlación está de acuerdo con los resultados de Ramos et ál. (50) en su estudio teórico de la influencia de campos eléctricos sobre puentes de hidrógeno en dímeros moleculares neutros y cargados. Los agregados con etanol y metanol, son sistemas neutros que, al igual que los estudiados por Ramos et ál., muestran un desplazamiento gradual del hidrógeno del puente de hidrógeno hacia el centro de tal interacción, acompañado por una contracción del sistema a medida que se fortalece esta interacción. En el presente estudio, el fortalecimiento de los puentes de hidrógeno se debe al aumento en el tamaño del agregado que da lugar a la presencia de efectos cooperativos. Como se explicará más adelante, se pueden entender los efectos cooperativos en términos del cambio de la polarizabilidad de las moléculas en forma similar a lo observado por Ramos et ál., quienes no variaron el tamaño de los agregados pero afectaron la polarizabilidad de los mismos con campos eléctricos.
Otros dos índices empleados para describir los efectos cooperativos son el corrimiento al rojo de las frecuencias de estiramiento de los enlaces O-H en las moléculas dadoras de protón (Δvd)yel factor de cooperatividad Ab para cada puente de hidrógeno primario. Por tanto, en este estudio, la existencia de cooperatividad también se refleja en el aumento en el corrimiento al rojo de la frecuencia de estiramiento del enlace O-H de las moléculas dadoras de protón en los puentes de hidrógeno de los heterotetrámeros y tetrámeros frente al corrimiento al rojo observado en la formación de un solo puente de hidrógeno entre dos moléculas (ver Tablas 1 y 2). El corrimiento al rojo asociado a la formación de puentes de hidrógeno con la molécula de agua como dadora de protón fue calculado teniendo en cuenta los dos modos de estiramiento: simétrico y asimétrico de sus dos enlaces O-H, ya que Sauer et ál. (51) mostraron que el mejor acuerdo entre el corrimiento al rojo calculado y el experimental para el dímero de agua se halla cuando el promedio para los dos modos de estiramiento se tiene en cuenta en el monómero aislado. En particular, la capacidad del funcional B3LYP para predecir dicho corrimiento al rojo fue evaluada por Koch y Houlthausen (52), quienes encontraron un excelente acuerdo entre los resultados con MP2 y los funcionales BLYP y B3LYP.
Finalmente, por medio del análisis del factor de cooperatividad se puede conocer cuantitativamente cuánto fue el reforzamiento (si es mayor que uno) o debilitamiento (si es menor que uno) de los puentes de hidrógeno en los agregados de cuatro moléculas debido a la formación de más de un puente de hidrógeno. Para los agregados mostrados en la Tabla 2, este parámetro está en el rango de 2,2 a 3.1 para el patrón geométrico (a), 0,8 a 2,0 para el patrón geométrico (b), 1,0 a 3.2 para el patrón geométrico (c), 1,2 a 2.3 para el patrón geométrico (d) y 1,3 a 1,9 para el patrón geométrico (e) (ver Tabla 2). Todos los puentes de hidrógeno son favorecidos desde los efectos cooperativos (Ab>1) con excepción de los puentes de hidrógeno C-H---A y C-H--- D en el heterotetrámero (etanol)3-agua (c) (Ab = 1) y el puente de hidrógeno C-H--- A en el heterotetrámero (etanol)3-agua(b) (Ab<1). (Ver Figura 5, donde se indica para estos puentes de hidrógeno sus distancias O--- O y elongaciones de sus enlaces O-H). Como se predice desde sus correspondientes factores de cooperatividad, los valores de estos índices geométricos de cooperatividad son muy similares a lo observado en los puentes de hidrógeno de los agregados de dos moléculas, debido al no reforzamiento o debilitamiento desde los efectos cooperativos. De modo que aunque de manera global todos los agregados obtienen una estabilización desde los efectos cooperativos, en algunos puentes de hidrógeno los efectos cooperativos positivos y negativos se cancelan mutuamente, o incluso prevalecen localmente los efectos negativos.
El análisis de estos índices de cooperatividad está de acuerdo con una explicación de tales efectos teniendo en cuenta tanto el cambio en la polarizabilidad de las moléculas involucradas en la formación de los puentes de hidrógeno, como de su comportamiento ácido/base. Un desplazamiento en las posiciones nucleares causa una redistribución de la densidad electrónica en cada uno de los sistemas que da lugar a un cambio en la polarizabilidad de las moléculas. Por ejemplo, se puede considerar un efecto negativo sobre la capacidad aceptora de protón de la molécula de agua en las estructuras con patrón geométrico (b), ya que esta, al actuar como doblemente aceptora de protón, ubica dos dadores de protón que representan dipolos aproximadamente paralelos que se repelen entre sí, como ha sido reportado por Steiner (14). Contrariamente, el carácter ácido de la molécula de agua en dicho patrón geométrico crece cuando la molécula se comporta como doblemente aceptora de protón, de acuerdo con el esquema ácido-base que Mó et ál. (33) emplearon en el análisis de la estabilidad relativa de los diferentes puentes de hidrógeno en trímeros de agua.
Como se observó desde el análisis termodinámico de los diferentes patrones geométricos, se favorecen principalmente las estructuras con el patrón geométrico (a) (ver Figuras 1 a 4), donde el carácter ácido/base de todas las moléculas se refuerzan mutuamente al ser todas las moléculas dadoras y aceptoras de protón, y a la vez la disposición cíclica también contribuye a la estabilización de estos agregados, ya que cada par de puentes de hidrógeno opuestos pueden representar dipolos antiparalelos atrayéndose los unos a los otros. De modo que aunque en el patrón geométrico (e), el puente de hidrógeno primario B-H---C, las moléculas B y C actúan como dadoras y aceptoras de protón al mismo tiempo, la cooperatividad no es tan alta como en los puentes de hidrógeno de las estructuras con patrón geométrico (a) (ver Ab en la Tabla 2), lo cual se puede deber a que en el patrón geométrico (e) por ser casi lineal no se da lugar a la estabilización desde la atracción de dipolos antiparalelos. Las fortalezas de los tres puentes de hidrógeno formando patrones cíclicos en los patrones geométricos (b), (c) y (d), se ven afectadas por la influencia de un cuarto puente de hidrógeno que refuerza uno de los puentes de hidrógeno dentro del patrón cíclico, mientras que debilita otro de los puentes de hidrógeno (ver Figuras 1, 2, 3, 4 y 5).
CONCLUSIONES
Por medio del análisis de parámetros termodinámicos y geométricos se observó la existencia de efectos cooperativos para todos los agregados. Dichos efectos pueden ser explicados cualitativamente en términos de la polarizabilidad y el carácter ácido-base de las moléculas entre las cuales se forman puentes de hidrógeno primarios.
La mayor cooperatividad la presentan los agregados con el patrón geométrico cíclico (a), donde las cuatro moléculas que los conforman actúan como dadoras y aceptoras de protón simultáneamente en cuatro puentes de hidrógeno primarios lineales. En algunos casos el incremento en la fuerza de algunos puentes de hidrógeno es tal, que los ubican como puentes de hidrógeno de fuerza moderada con la contribución principal desde la energía electrostática, y por tanto tienen menos aporte de la energía de dispersión, como semuestra enlasegundatabladelartículo de revisión para puentes de hidrógeno en el estado sólido de Steiner (14). Es posible que con un aumento mayor en el tamaño de los agregados, los puentes de hidrógeno secundarios se refuercen cada vez más, de modo que presenten algún efecto apreciable en la estabilidad termodinámica de los agregados.
La comparación de los heterotetrámeros(etanol)3-aguaconlosotrosagregados caracterizados en este estudio, muestra una gran similitud en todos los parámetros analizados, lo que indica que los efectos cooperativos son fuertemente dependientes del patrón geométrico adoptado por los agregados, más que de la naturaleza de los monómeros que los conforman. Este comportamiento sugiere que este tamaño para los agregados aún no puede proveer suficiente información a nivel molecular que pudiera ser entendida en términos del por qué se forma y es tan estable el azeótropo etanol-agua, mientras que el metanol no forma esta mezcla no ideal, a pesar de ser un alcohol alifático primario, al igual que el etanol.
AGRADECIMIENTOS
Los autores expresan sus agradecimientos a la Universidad de Antioquia por la financiación del programa Sostenibilidad. Juan F. Espinal y Sol M. Mejía agradecen a Colciencias y a la Universidad de Antioquia por las becas doctorales. Los autores agradecen a uno de los evaluadores la sugerencia sobre la correlación de la distancia O-H con la distancia O---O.
REFERENCIAS BIBLIOGRÁFICAS
1. DeMatteo, M. P.; Snyder, N. L.; Morton, M.; Baldisseri, D. M.; Hadad, C. M.; Peczuh, M. W. Septano-se carbohydrates: Synthesis and conformational studies of methyl a-D-glycero-D-idoseptanoside and methyl ß-D-glycero-D-guloseptano-side. J. Org. Chem. 2005 . 70(1): 24-38. [ Links ]
2. Fonseca G. C.; Bickelhaupt F. M.; Snijders J. G.; Baerends E. J. Hydrogen bonding in DNA base pairs: Reconciliation of theory and experiment. J. Am. Chem. Soc. 2000. 122 (17): 4117-4128. [ Links ]
3. Mingos, D. M. P. Supramolecular assembly via hydrogen bonds. Washington, D.C.: Springer. 2004. p. 33. [ Links ]
4. Rozas, I. On the nature of hydrogen bonds: an overview on computational studies and a word about patterns. Phys. Chem. Chem. Phys. 2007. 9(22): 2782-2790. " [ Links ]
5. Lutz, H. D. Structure and strengthof hydrogen bonds in inorganic solids. J. Mol. Struct. 2003 . 646(1-3): 227-236. [ Links ]
6. Curtiss, L. A.; Blander, M. Ther-modynamic properties of gas-phase hydrogen-bondedcomplexes. Chem. Rev. 1988. 88(6): 827-841. [ Links ]
7. Fernández, R.; Japas, M. L. Critical behavior of fluid binary mixtures: intermolecular parameters and ther-modynamic properties. J. Phys. Chem. 1992. 96(12): 5115-5121.' [ Links ]
8. Travers, F.; Douzou, P. Dielectric constants of alcoholic-water mixtures at low temperatura. J. Phys. Chem. 1970. 74(10): 2243-2244.' [ Links ]
9. Montoya, M. I.; Quintero, J. A.; Sánchez, O. J.; Cardona, C. A. Evaluación económica del proceso de obtención de alcohol carburante a partir de caña de azúcar y maíz. Rev. Univ. EAFIT. 2005.41(139): 76-87. [ Links ]
10. Westgate, P. J.; Ladisch, M. R. Sorption of organics and water on starch. Ind. Eng. Chem. Res. 1993. 32(8): 1676-1680. [ Links ]
11. Hobza, P.; Havlas, Z. Blue-Shifting hydrogen bonds. Chem. Rev. 2000. 100(11): 4253-4264. [ Links ]
12. Scheiner, S.; Kar, T. Red- versus blue-Shifting hydrogen bonds: are there fundamental distinctions? J. Phys. Chem. A. 2002. 106(9): 1784-1789. [ Links ]
13. Desiraju, G. R. Hydrogen bridges in crystal engineering: interactions without borders. Acc. Chem. Res. 2002. 35(7): 565-573. [ Links ]
14. Steiner T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002. 41(1): 48-76. [ Links ]
15. Coussan, S.; Roubin, P.; Perchard, J. P. Hydrogen bonding in ROH:R'OH (R, R' = H, CH3, CH2CH5) Heterodimers: Matrix-Dependent structure and infrared-induced isomerization. J. Phys. Chem. A. 2004. 108(35): 7331-7338. [ Links ]
16. Katrib, Y.; Mirabel, P.; Le Calve, S.; Week, G.; Kochanski, E. Experimental uptake study of ethanol by water droplets and its theoretical modeling of cluster formation at the interface. J. Phys. Chem. B. 2002. 106(29): 7237-7245. [ Links ]
17. Oliveira, B. G.; Vasconcellos, M. L. A. A. Hydrogen bonds in alcohols: water complexes: a theoretical study about new intramolecular interactions via CHELPG and AIM Calculations. J. Mol. Struct: THEOCHEM 2006. 774 (1-3): 83-88. [ Links ]
18. van Erp, T. S.; Meijer, E. J. Ab Initio molecular dynamics study of aqueous solvation of ethanol and ethylene. J. Chem. Phys. 2003. 118(19): 8831-8840. [ Links ]
19. Masella, M.; Flament, J. P. Relation between cooperative effects in cyclic water, methanol/water, and methanol trimers and hydrogen bonds in methanol/water, ethanol/water, and dimethylether/Water Heterodimers. J. Chem. Phys. 1998. 108(17): 7141-7151. [ Links ]
20. Mejia, S. M.; Espinal, J. F.; Restrepo, A.; Mondragon, F. Molecular interaction of (ethanol)2-water heterotrimers. J. Phys. Chem. A. 2007. 111(33): 8250-8256. [ Links ]
21. Grabowski, S. J. Ab Initio calculations on conventional and unconventional hydrogen bonds-study of the hydrogen bond strength. J. Phys. Chem. A 2001. 105(47): 10739-10746. [ Links ]
22. Parthasarathi, R.; Subramanian. V.; Sathyamurthy, N. Hydrogen bonding in phenol, water, and phenol-water clusters. J. Phys. Chem. A. 2005. 109(5): 843-850. [ Links ]
23. González, L.; Mó, O.; Yáñez, M.; Elguero J. Cooperative effects in water trimers. The performance of density functional approaches. J. Mol. Struct: THEOCHEM. 1996. 371: 1-10. [ Links ]
24. Xantheas, S. S.; Dunning, T. H. Ab initio studies of cyclic water cluster (H2O)n, n=1-6. I. Optimal structures and vibrational Spectra. J. Chem. Phys. 1993. 99(11): 8774-8792. [ Links ]
25. González, L.; Mó, O.; Yáñez, M. Density functional theory study on ethanol dimers and cyclic ethanol trimers. J. Chem. Phys. 1999. 111(9): 3855-3861. [ Links ]
26. Mandado, M.; Graña, A. M.; Mosquera, R. A. On the structures of the methanol trimer and their cooperative effects. Chem. Phys. Lett. 2003. 381(1-2): 22-29. [ Links ]
27. Masella, M.; Flament, J.-P. A Many-Body model for alcohols: applications to the cyclic methanol/water hetero trimers, and to the (methanol)n, (ethanol)n and (t-butanol)n cyclic clusters (n = 2-6). Mol. Phys. 1998. 95(1): 97-106. [ Links ]
28. González, L.; Mó, O.; Yáñez, M. High level ab initio and density functional theory studies on methanol-water dimers and cyclic methanol(water)2 trimer. J. Chem. Phys. 1998. 109(1): 139-150. [ Links ]
29. Boyd, S. L.; Boyd, R. J. A. Density functional study of methanol clusters. J. Chem. Theory Comput. 2007. 3(1): 54-61. [ Links ]
30. Glendening, E. D. Natural energy decomposition analysis: extension to density functional methods and analysis of cooperative effects in water clusters. J. Phys. Chem. A. 2005. 109(51): 11936-11940. [ Links ]
31. Umeyama, H.; Morokuma, K. The origin of hydrogen bonding. An energy decomposition study. J. Am. Chem. Soc. 1977.99(5): 1316-1332. [ Links ]
32. Pérez, J. F.; Hadad, C. Z.; Restrepo, A. Structural studies ofthe water tetramer. Int. J. Quantum Chem. 2008. 108(10): 1653-1659. [ Links ]
33. Mó, O.; Yáñez, M.; Elguero, J. Cooperative (nonpairwise) effects in water trimers: an ab initio molecular orbital study. J. Chem. Phys. 1992. 97(9): 6628-6638. [ Links ]
34. Mejía, S. M.; Espinal, J. F.; Mondragón, F. Estudio del azeótropo etanol-agua. Caracterización molecular de dímeros de etanol, heterodímeros y heterotrímeros de etanol-agua. Energética 2006. (36): 5-12. [ Links ]
35. Johnson, E. R.; Dilabio, G. A. Structure and Binding Energies in van der Waals dimers: comparison between density functional theory and correlated ab initio methods. Chem. Phys. Lett. 2006. 419(4-6): 333-339. [ Links ]
36. Rozas, I.; Alkorta, I.; Elguero, J. Unusual hydrogen bonds: H---p interactions. J. Phys. Chem. A. 1997. 101(49): 9457-9463. [ Links ]
37. Jursic, B. S. Study of the water-methanol dimer with gaussian and complete basis set ab initio, and density functional theory methods. J. Mol. Struct. THEOCHEM. 1999. 466(1): 203-209. [ Links ]
38. van Duijneveldt, F. B.; van Duijne-veldt-vande Rijdt, J. G. C. M.; van Lenthe, J. H. State of the art in counterpoise theory. Chem. Rev. 1994. 94(7): 1873-1885. [ Links ]
39. Kar, T.; Scheiner, S. Comparison of cooperativity in CH---O and OH---O hydrogen bonds. J. Phys. Chem. A. 2004. 108(42): 9161-9168. [ Links ]
40. Huyskens, P. L. Factors governing the influence of a first hydrogen bond on the formation of a second one by the same molecule or ion. J. Am. Chem. Soc. 1977. 99(8): 2578-2582. [ Links ]
41. Haulalt, M. C.; Huyskens, P. L. Influence of a first hydrogen bond on the formation of a second one by alkyl- or dialkylammonium ions. The J. Phys. Chem. 1975. 79(17): 1812-1816. [ Links ]
42. Weibel, J. D.; Jackels, C. F.; Swofford, R. L. Experimental and ab initio investigation of the O-H overtone vibration in etanol. J. Chem. Phys. 2002. 117(9): 4245-4254. [ Links ]
43. Coussan, S.; Bouteiller, Y.; Perchard, J. P.; Zheng, W. Q. Rotational isomerism of ethanol and matrix isolation infrared spectroscopy. J. Phys. Chem. A. 1998. 102(29): 5789-5793. [ Links ]
44. Fang, H. L.; Swofford, R. L. Molecular conformers in gas-phase ethanol: a temperature study of vibrational overtones. Chem. Phys. Lett. 1984. 105(1): 5-11. [ Links ]
45. Kakar, R. K.; Quade, C. R. Microwave rotational spectrum and internal rotation in gauche ethyl alcohol. J. Chem. Phys. 1980. 72(8): 4300-4307. [ Links ]
46. Frurip, D. J.; Curtiss, L. A.; Blander, M. Thermal conductivity measurements and molecular association in a series ofalcohols vapors: methanol, ethanol, isopropanol, and t-Butanol. Int. J. Thermophys. 1981. 2(2): 115-132. [ Links ]
47. Barrow, G. M. Heat Capacity, Gas Imperfection, infrared spectra, and internal rotation barriers of ethyl alcohol. J. Chem. Phys. 1952. 20(11): 1739-1744. [ Links ]
48. Kretschmer, C. B.; Wiebe, R. Pressure-Volume-Temperature relationships of alcohol vapors. J. Am. Chem. Soc. 1954. 76(9): 2579-2583. [ Links ]
49. Weltner, W.; Pitzer, K. S. Methyl Alcohol: The entropy, heat capacity and polymerization equilibria in the vapor, and potential barrier to internal rotation. J. Am. Chem. Soc. 1951. 73(6): 2606-2611. [ Links ]
50. Ramos, M.; Alkorta, I.; Elguero, J.; Golubev, N. S.; Denisov, G. S.; Benedict, H.; Limbach, H. Theoretical study of the influence of electric fields on hydrogen-bonded acid-base complexes. J. Phys. Chem. A. 1997. 101(50): 9791-9800. [ Links ]
51. Sauer, J.; Ugliengo, P.; Garrone, E.; Saunders, V. R. Theoretical study of van der Waals complexes at surface sites in comparison with the experiment. Chem. Rev. 1994. 94(7): 2095-2160. [ Links ]
52. Koch, W.; Holthausen M. C. Hydrogen bonds and weakly bound systems. In: A chemist's guide to density functional theory. New York: Wiley-Vch. 1999. pp. 217-236. [ Links ]

