










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804On-line version ISSN 2357-3791
Rev.Colomb.Quim. vol.38 no.1 Bogotá Jan./Apr. 2009
ISOMERIZACIÓN DEL 1-CICLOHEXILOCTANO EN ZEOLITAS H-ZSM-22 Y H-Y
1-CYCLOHEXYLOCTANE ISOMERIZATIONINTO H-ZSM-22 AND H-YZEOLITES
ISOMERIZACÁO DE 1-CICLOHEXILOCTANO NA ZEÓLITAS H-ZSM-22 YH-Y
Jorge Quintero S.1, Cristian Blanco T.1,2
1 Escuela de Química, Universidad Industrial de Santander, Bucaramanga, Colombia.
Recibido: 07/11/08 - Aceptado: 13/04/09
RESUMEN
En este trabajo se presentan los resultados de la evaluación energética de los intermediarios de la isomerización del 1-ciclohexiloctano en dos etapas. La primera etapa corresponde a la isomerización del 1-ciclohexiloctano en fase gaseosa vía carbocationes clásicos, usando el nivel de teoría DFT/B3LYP y conjunto de funciones base 6-31G. En la segunda etapa se desarrolló la coordenada de reacción de la isomerización del 1-ciclohexiloctano en presencia de zeolitas acidas H-ZSM-22 y H-Y empleando el método ONIOM, usando los niveles de teoría B3LYP/ 6-31G:UFF. Los resultados muestran diferencias significativas entre las reacciones en fase gaseosa y condensada. Estas diferencias ponen de manifiesto la influencia del confinamiento cuántico que sufren las moléculas al interior de los sistemas microporosos del tipo zeolitas.
Palabras clave: zeolitas, isomerización, selectividad, química cuántica, mecánica molecular.
ABSTRACT
We studied the isomerization mechanism of 1-cycle-hexyloctane either in gas phase or inside zeolites using quantum models. In the gas phase the reaction was simulated following the classical scheme via carbocations. On the other hand, the isomerization reaction was modeled with 1-cycle-hexyloctane inside HZSM-22 and H-Y acid zeolites (condensed phase). We used the ONIOM(B3LYP/6-31G: UFF) hybrid method to study such behavior in both zeolites. Our results show that the reaction follows different pathways when the simulations are performed either in gas phase or within the zeolites pores. Such differences show the importance of quantum confinement inside microporous materials.
Key words: zeolites, isomerization reaction, selectivity, quantum chemistry, molecular mechanic.
RESUMO
Neste trabalho foram desenvolvidos os mecanismos das reacção de isomerização de 1-ciclo-hexil-octano através de duas etapas. Na primeira etapa, a reação foi realizada como carbocátions clasicos, utilizando-se o método DFT/B3LYP e as funções básicas 6-31G. Na segunda, a reação de isomerização de 1-ciclo-he-xil-octano foi estudou em zeolites ácido H-ZSM-22 e H-Y utilizando-se o método híbrido ONIOM o nível B3LYP e funções básicas 6-31G.
Palavras-chave: zeólitas, reação de isomerização, seletividade, química quàntica, mecânica molecular.
INTRODUCCIÓN
Las aplicaciones industriales de las zeolitas son consecuencia de sus propiedades físico-químicas. Muchas de estas propiedades resultan tan ventajosas que han motivado la sustitución de otros materiales convencionales por zeolitas en numerosos procesos industriales como la separación (1), intercambio iónico (2) y en procesos catalíticos como la isomerización (3) y craqueo de hidrocarburos (4).
La conversión de hidrocarburos es controlado por etapas de deshidrogenación, transporte de las olefinas hasta los sitios ácidos de Bronsted, isomerización o craqueo de los hidrocarburos, quimiodesorción y formación de nuevas olefinas, e hidrogenación de las olefinas en los sitios metálicos (5).
En los procesos de isomerización existen evidencias experimentales relacionadas con la actividad catalítica de las zeolitas, en casos en que las moléculas no alcanzan a penetrar totalmente los canales del poro, conocida como catálisis en la boca del poro (6). Jacobs et ál. estudiaron la conversión del 1-ciclohexiloctano usando zeolitas bifuncionales Pt/H-ZSM-22 (7) y Pt/H-Y (8). Los productos mayoritarios en ambos materiales son los de isomerización. Sin embargo, en las zeolitas Pt/H-Y se evidencia la formación de isómeros ciclohexil 5a y ciclopentil 4a dialquilsustituídos sobre la cadena alifática; mientras que en las zeolitas Pt/H-ZSM-22 únicamente se observa la formación de las últimas especies mencionadas 4a (Figura 1).
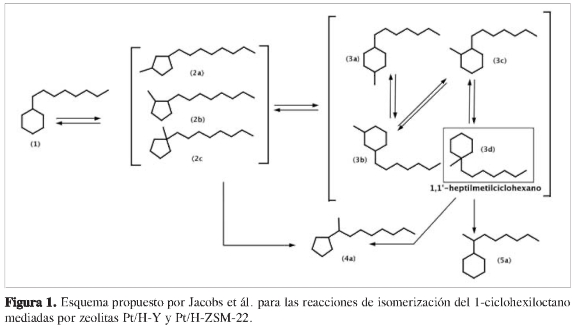
También se destaca el hecho de que los isómeros dialquil-ciclohexil sustituidos únicamente se forman a partir del 1-heptil-1 '-metilciclohexano 3d. Por tanto, la única posibilidad de obtener estos isómeros es a través de la transformación del anillo hexacíclico vía intermedio alcóxido, que ocurre en la boca del poro, según lo plantean Jacobs et ál. (7, 8).
A pesar de que el modelo de catálisis en la boca del poro describe adecuadamente el fenómeno de selectividad, existe una controversia en torno a este planteamiento, debido a que la superficie externa de las zeolitas corresponde a 0.5% de la superficie total del material (9). Esta polémica es causada en parte por la dificultad para observar experimentalmente los procesos mediados por la superficie de la zeolita y la falta de una visión microscópica y atomística detallada del material.
En el presente trabajo se llevó a cabo una serie de simulaciones para obtener los perfiles de energía correspondientes a los mecanismos de isomerización del 1-ciclohexiloctano controlada por zeolitas ácidas H-ZSM-22 y H-Y. Adicionalmente, se calcularon los perfiles energéticos del mismo proceso de isomerización en fase gaseosa.
MATERIALES Y MÉTODOS
Coordenada de reacción en fase gaseosa
Las simulaciones se iniciaron a partir de la formación del carbocatión 1- ciclohexiloctano. Los pasos siguientes en la isomerización del 1- ciclohexiloctano conducen a la formación del 1-heptil-í-4-metilciclohexano (IV) y 1-heptil-1'-metilheptilciclohexano (X) representados en la Figura 2.
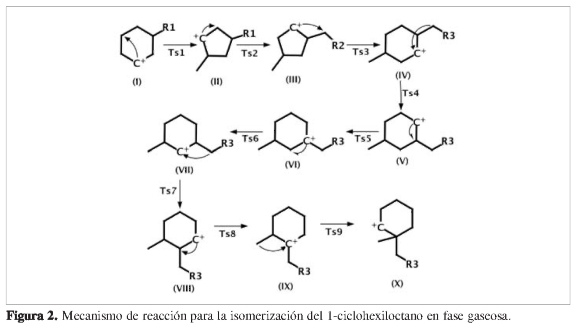
Las geometrías moleculares tanto para reactivos como productos se optimizaron localizando puntos críticos de menor energía dentro de la superficie de energía potencial, utilizando el nivel de teoría B3LYP (10) y conjuntos de funciones de base 6-31G (11). Para encontrar el estado de transición que conecta a los reactivos y a los productos, de cada etapa de la reacción, se utilizó el método Quasi-Newton y Transito Sincrónico (12). Estos intermedios se confirmaron mediante el método coordenada de reacción intrínseca (13). Finalmente se analizaron los modos vibracionales a través de la diagonalización de la matriz hessiana, teniendo en cuenta que la aparición de una frecuencia negativa confirma la obtención de un estado de transición.
Coordenada de reacción en fase condensada
Inicialmente se obtuvieron las estructuras más estables para las zeolitas ácidas H-ZSM-22 y H-Y con base en los parámetros geométricos de la unidad asimétrica y el grupo espacial al cual pertenece cada material. En el caso de la zeolita H-ZSM-22, se construyó una celda unitaria, con fórmula Si138AlO330H104 y grupo espacial Cmcm cúbica, que contiene canales unidimensionales constituida por anillos de 10 miembros en dirección [001], con dimensiones de 5,5 x 4,5 Å. Los parámetros de celda fueron a = 13,86 Å,b = 17,41 Å y c = 5,04 Å(14).
Para la zeolita H-Y, que pertenece a la familia de la Faujasita, se construyó una celda unitaria con fórmula Si147Al45O440H160 y grupo espacial Fd3m, constituida por cajas de sodalitas cubo-octahédricas. Estas estructuras contienen grandes cavidades denominadas supercajas, las cuales se conectan tetrahédricamente por canales de 12 miembros. Los parámetros de celda fueron a=b=c=24,76 Å(15).
Mecanismo de reacción en fase condensada
Numerosos estudios experimentales (16) y teóricos (17) indican que la etapa correspondiente a la isomerización de hidrocarburos se logra a partir de la formación de grupos alcóxidos generados por el transporte de olefinas previamente formadas en los sitios metálicos de la zeolita. En este caso, las simulaciones se iniciaron localizando un sitio ácido de Brensted en la boca de la superficie externa de la zeolita, mediante la construcción de un enlace C-O del sistema zeolita-huésped, donde el anillo hidrocarbonado fue ubicado sobre la boca del poro y la cadena alifática fue orientada hacia la parte interna del canal de la zeolita. Debido a la posibilidad de iniciar la reacción en cualquiera de los dos oxígenos del grupo alcóxido, las simulaciones se iniciaron desde dos configuraciones diferentes, proporcionando dos rutas de isomerización independientes.
Para el desarrollo de la coordenada de reacción en fase condensada se empleó el método ONIOM. Las simulaciones se llevaron a cabo en sistemas constituidos por 573 átomos para la zeolita H-ZSM-22 y 792 átomos para la zeolita H-Y. Las geometrías moleculares, de cada uno de los intermedios y de los estados de transición (ver Figura 3), se optimizaron utilizando el método ONIOM2 (18). En la capa de bajo nivel, donde sólo se consideran efectos de campo, se utilizó el campo de fuerza universal (19). En la capa de alto nivel de teoría, conformada por la cadena hidrocarbonada y un fragmento de la zeolita (OH)-AlO2Si2, donde ocurre la ruptura y formación de enlaces, se utilizó el funcional B3LYP (10) y el conjunto de base 6-31G, implementados en el programa Gaussian 03 (20).
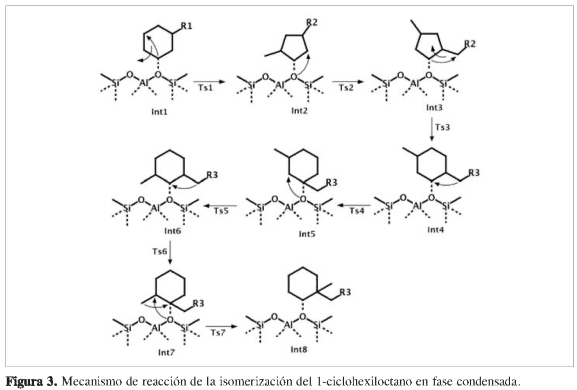
La geometría molecular de cada estado de transición se optimizó mediante la construcción de diferentes configuraciones que conectan los estados de los reactivos y de los productos en cada etapa de la coordenada de reacción. Para lograr este objetivo se utilizo el método de gradiente analítico de Berny (21). Posteriormente se analizaron los modos vibracionales del estado de transición mediante la diagonalización de la matriz Hessiana, teniendo en cuenta la aparición de una frecuencia negativa, de igual manera como se obtuvieron estos estados en los estudios de la reacción en fase gaseosa.
RESULTADOS Y DISCUSIÓN
Fase gaseosa
En la Figura 4 se muestra el perfil de energía de la reacción en fase gaseosa. Se observa que los valores de energía relativa para aductos cis y trans, en cada etapa de la coordenada de reacción, son muy cercanos. Nuestros resultados muestran que no hay formación preferencial de aductos cis o trans. Sin embargo, los intermedios catiónicos VI-(cis,trans)y IX-(cis,trans) aparecen como los más estables de la reacción total.
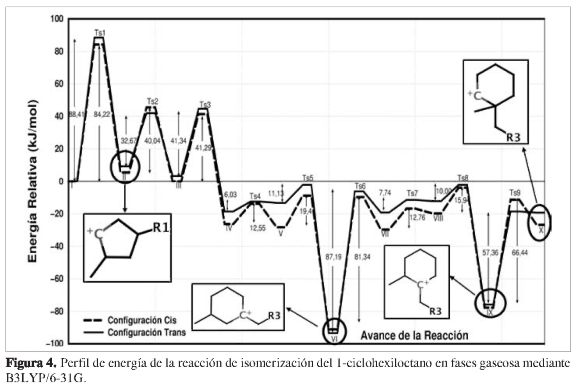
La primera etapa de la reacción se caracterizó por la contracción del anillo hexacíclico para formar los intermedios catiónicos ciclopentil di-sustituidos II-(cis, trans), a través de estados de transición bicíclicos cuasi-triangular Ts1-(cis, trans) debido a la ruptura del enlace C4-C2 y la transferencia del hidruro H4 entre los carbonos C2 y C4. La energía de activación para el Tsl-cis y Tsl-trans fue de 84,22 kJ/mol y de 88,41 kJ/mol, respectivamente. Estos valores corresponden a la primera etapa lenta de la reacción total.
Los intermedios catiónicos II-(cis, trans) se modifican rápidamente mediante la transferencia del hidruro H3 desde C3 hacia el C2, a través de estados de transición Ts2-(cis,trans), para la obtención de los intermedios catiónicos III-(cis,trans). Posteriormente, los intermedios III-(cis,trans) sufren una etapa de expansión del anillo ciclopentilo a través de los estados de transición Ts3-(cis,trans), dirigidos por el reordenamiento concertado de la ruptura del enlace C2-C3, transferencia del hidruro H4 desde el C4 hacia el C2 y la formación del enlace entre C2 y C4. Aquí se observa nuevamente el re-arreglo bicíclico cuasi-triangular entre el anillo ciclopentil y la cadena alifática que se dirige hacia el carbono C2.
Las energías de activación fueron 41,29 kJ/mol para el Ts3-cis y 41,34 kJ/mol para el Ts3-trans. Posteriormente se obtienen los intermedios catiónicos IV-cis y IV-trans con energías relativas -26,32 kJ/mol y -18,54 kJ/mol, menores que el intermedio de partida. Esta estabilidad es favorecida por la proximidad entre el carbocatión secundario y los grupos metilo y n-heptilo.
Sin embargo, el perfil de energía muestra dos intermedios catiónicos mucho más estables energéticamente. Es el caso de los isómeros VI-(cis,trans)y lX-(cis, trans). Los isómeros VI-(cis, trans) son formados mediante etapas de transferencia del grupo n-heptilo debido a la ruptura del enlace C4-C3 y la formación del enlace C4-C2 con energía de activación de 12,55 kJ/mol para el Ts4-cis y 6,03 kJ/mol para el Ts4-trans, a través de la formación de los intermedios V-(cis, trans), los cuales transfieren un hidruro H4 desde el C3 hacia el C2 y producen los intermedios catiónicos VI-(cis,trans). Estos intermedios se caracterizan por la deficiencia de carga localizada sobre el carbono terciario adyacente al grupo n-heptilo, altamente estable.
Finalmente se obtuvieron los intermedios X-(cis,trans) a partir de la migración del grupo metilo hacia el carbono elec-trodeficiente del fragmento cíclico (IX-Ts9-X) a través de estados de transición Ts9-(cis,trans) cuyas energías de activación fueron 66,44 kJ/mol y 57,36 kJ/mol, respectivamente. Este paso se constituye como la tercera etapa lenta de la reacción global. Estos últimos intermedios corresponden al isómero 1,1'-heptil-metilciclohexano, clave para la formación de especies dialquil- sustituidos con fragmentos ciclopentil y ciclohexil.
Fase condensada
En general, las reacciones de isomerización del 1-ciclohexiloctano, catalizadas por zeolitas H-ZSM-22 y H-Y, se desarrollaron teniendo en cuenta el esquema de reacción representado en la Figura 3.
Isomerización del 1-ciclohexiloctano mediada por la zeolita H-ZSM-22
Los dos perfiles de energía de la reacción de isomerización del 1-ciclohexiloctano en la zeolita H-ZSM-22 muestran un aumento progresivo de la energía relativa durante el avance de la reacción. Sin embargo, la ruta que sigue la configuración A permite la obtención de los intermedios Int4-cis e Int4-trans. Con la configuración B, la primera etapa, que corresponde a la formación de los intermedios Int2-cis e Int2-trans, muestra energías de intermedios relativamente superiores, lo cual indica que esta ruta energética es menos favorable para la isomerización del 1-ciclohexiloctano.
En la configuración A, las energías relativas para los intermedios y estados de transición, en función del avance de reacción, revelan algunas etapas determinantes del proceso de isomerización del 1-ciclohexiloctano. Los resultados se muestran en la Figura 5.
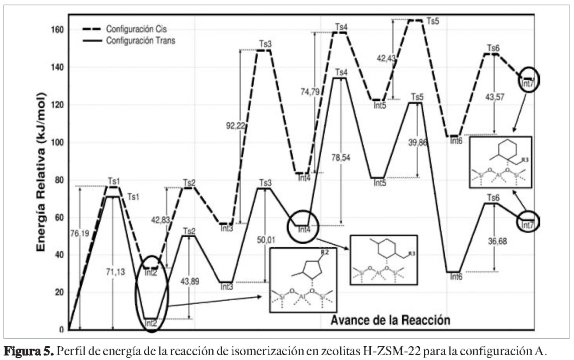
La primera etapa determinante corresponde a la contracción del anillo hexacíclico, lo cual produce dos estados de transición bicíclicos cuasi-triangulares de tres átomos de carbonos, según se muestra en la Figura 6. En los estados de transición Ts1-cis y Ts1-trans, el enlace C1-O1 se rompe hasta alcanzar una distancia de 1,660 Á, tanto para el Ts1-cis como para el Ts1-trans, necesario para que el anillo pentacíclico se reordenara de tal manera que se pudieran alcanzar los intermedios Int2-cis e Int2-trans.
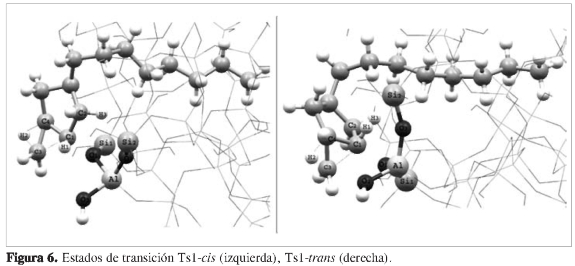
Simultáneamente ocurre la ruptura del enlace C1-C3, formación de un nuevo enlace C1-C4 y transposición de un hidruro H2 desde el carbono C4 hasta el carbono C3. Para el estado de transición Ts1-cis, cuya energía de activación fue de 76,19 kJ/mol, el carbono C3 se orientó por encima del plano del anillo pentacíclico.
Para el Ts1-íra«s, cuya energía de activación fue de 71,13 kJ/mol, el carbono C3 se orientó por debajo del plano del anillo pentacíclico.
En los intermedios Int2-ds e Int2-trans, tanto el anillo pentacíclico como la cadena alifática sufren rotaciones para orientar el carbono electrodeficiente, de tal manera que puedan ser estabilizados por el oxígeno de la zeolita.
Otras reacciones que se presentaron fueron generadas entre los intermedios Int2-(cis,trans) e Int3-(cis,trans), a través de los estados de transición Ts2-cis y Ts2-trans. El hidruro H3 es transferido desde el carbono C1 hacia el carbono C2. Adicionalmente, en el C1-O1 se rompe y se forma nuevamente entre C2-O1. Tanto para aductos cis como trans, la configuración de las estructuras se caracterizó por la cercanía de los átomos C1 y C2 hacia el átomo O1.
Parte del anillo es desplazado hacia el exterior de la boca de poro y orientado de tal manera que ambos carbonos electrodeficientes interaccionen fuertemente con el oxígeno de la zeolita para ser estabilizados. La energía de los estados de transición Ts2-ds y Ts2-trans fue de 42,83 kJ/moly 43,89 kJ/mol, respectivamente. Esta energía es mucho menor si se compara con la energía de los estados de transición Ts1-cis y Ts1-trans, lo cual indica que no es una etapa determinante de la reacción. Además, luego de alcanzar dichos estados de transición se obtienen los intermedios Int3-cis e Int3-trans,cuyas energías son mayores que los Int2-(cis,trans).
La segunda etapa determinante es la transposición de la cadena alifática entre los intermedios Int4-(cis,trans)e Int5-(ris,trans). El átomo C3, que inicialmente estaba enlazado al carbono C4 con una distancia de enlace de 1,541 Å para el Int4-ris y 1,540 Å para el Int4-trans, se rompe y se dirige simultáneamente hacia el carbono C1 alcanzando los estados de transición Ts4-cis y Ts4-trans, cuyas energías relativas fueron 74,79 kJ/mol y 78,54 kJ/mol. En los estados de transición, nuevamente se observó el reordenamiento bicíclico cuasi-triangular formado entre los carbonos C1-C4-C3 y la transposición del hidruro H11 desde el átomo C1 hacia el átomo C4. Tanto para el Ts4-cis como para el Ts4-trans fue necesario que el anillo pentacíclico se reordenara de tal manera que se lograran alcanzar los intermedios Int5-cis e Int5-trans.
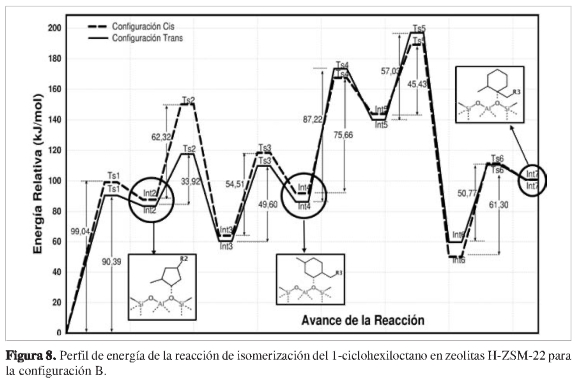
La etapa final de la reacción corresponde de nuevo a la transposición de la cadena alifática desde el carbono C4 hacia el carbono C1, con ruptura de los enlaces C4-C3 y C1-O1, y la formación de un nuevo enlace, C3-C1. Simultáneamente se transpone el hidruro H1 desde el átomo C1 hacia el átomo C3, mediante lo cual se obtiene un alcóxido terciario. Esto ocurre mediante estados de transición bicíclicos cuasi-triangular Ts6-ris y Ts6-trans (Figura 7), entre los átomos C1-C4-C3. Según el perfil de energía, se observó claramente una diferencia de energía de 75,23 kJ/mol entre los productos Int7-cis e Int7-trans, indicando la formación preferencial del Int7-trans.
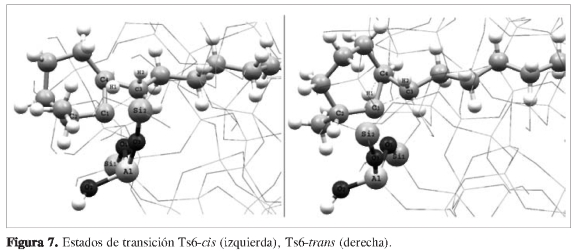
Para la configuración B, las energías relativas para los intermedios y estados de transición fueron representadas mediante un perfil de energía, como lo muestra la Figura 8. Los detalles para todos los intermedios y estados de transición se omiten. A medida que avanza la reacción, las energías relativas de los aductos cis y trans en cada intermedio fueron muy cercanas y aumentaron drásticamente. Es el caso de los intermedios Int2-(cis,trans)e Int5-(cis,trans). Este último, correspondió a la formación de un alcóxido terciario generado a partir de la transposición de la cadena alifática.
Sin embargo, los intermedios Int6-(cis,trans) disminuyen su energía relativa. Estas especies, como ya se ha mencionado, son generadas a través de estados de transición Ts5-(cis,trans) mediante transferencia de hidruro para formar alcóxidos secundarios. En dichos estados de transición, los átomos de carbonos C2 y C1 forman un reordenamiento triangular con el oxígeno de la zeolita debido a la deficiencia de carga causada por la transferencia del hidruro.
Finalmente, la reacción de isomerización termina con la formación de los productos Int7-cis e Intl-trans, cuyas energías relativas fueron 100,71 kJ/mol y 100,75 kJ/mol, respectivamente. Enam-bos productos se observaron energías relativas elevadas, en comparación con Int1, producidas por la orientación de la cadena alifática y la alta interacción de repulsión del sistema zeolita-huésped. Además, los alcóxidos formados presentan alto impedimento estérico provocado tanto por el anillo hexacíclico como por la cadena alifática. Al igual que en la configuración A, la formación de los intermedios Int8-(cis,trans) es completamente suprimida debido a la alta energía relativa para cada aducto (101,78 kJ/mol para cis y 68,54 kJ/mol para trans).
Isomerización del 1-ciclohexiloctano mediada por la zeolita H-Y
El avance de la reacción también presentó diferentes modos de reordenamiento estructural de cada uno de los intermedios de reacción: contracción y expansión de anillo hidrocarbonado, transposición de hidruro y de la cadena alifática.
Para la configuración A, las energías relativas se muestran en la Figura 9. Se observan dos puntos críticos con la mayor energía de activación, correspondientes a etapas determinantes de la reacción. El primero se localizó entre los intermedios Int1 e Int2-(cis,trans) a través de dos estados de transición cuyas energías relativas fueron 77,82 kJ/mol para el Ts1-cis y 72,09 kJ/mol para el Ts1-írans.
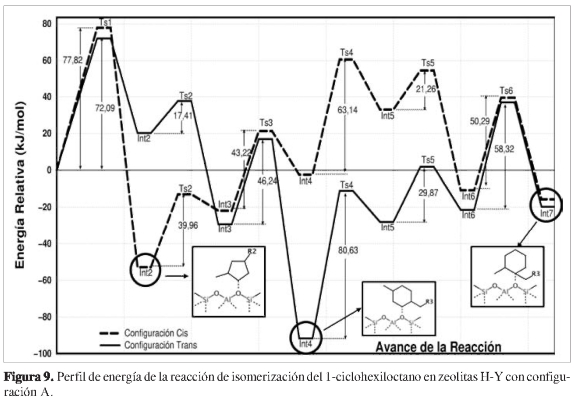
La primera etapa determinante se obtuvo debido a la contracción del anillo hexacíclico obteniendo dos estados de transición cuasi-triangulares de tres átomos de carbonos. La segunda etapa determinante fue localizada entre los intermedios Int4-(cis,trans) e Int5-(cis,trans) a través de los estados de transición Ts4-cis, con una energía relativa de 63,14 kJ/mol, y Ts4-trans con una energía relativa de 80,63 kJ/mol.
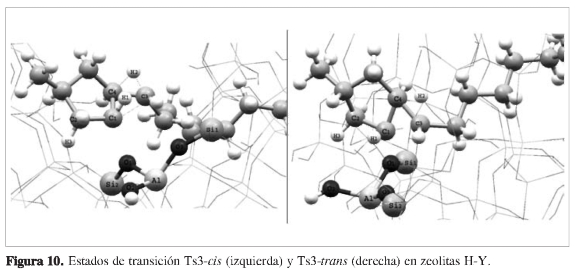
Adicionalmente, otras reacciones de modificación del hidrocarburo se presentaron entre los intermedios Int2-(cis, trans) e Int3-(cis,trans), mediante los estados de transición Ts2-cis y Ts2-trans, en los cuales se transfiere el hidruro H3 desde el carbono C1 hacia el carbono C2. El enlace C1-O1 se rompe y se forma uno nuevo entre C1-O1. Seguidamente el anillo pentacíclico se expande nuevamente para obtener los aductos hexacíclicos cis y trans a través de los estados de transición Ts3-cis y Ts3-trans (Figura 10), con energías relativas de 43,22 kJ/mol y 46,24 kJ/mol, respectivamente. El enlace C-O se rompe temporalmente permitiendo la formación de un nuevo enlace entre los átomos C1-C3.
La segunda etapa determinante es la transposición de la cadena alifática entre los intermedios Int4-(cis,trans) e Int5-(cis,trans). Simultáneamente el enlace C3-C4 se rompe y sucesivamente se forma un nuevo enlace C3-C1, alcanzando los estados de transición Ts4-cis y Ts4-trans a través de la ruptura del enlace C1-O1. Nuevamente se observó el reordenamiento bicíclico cuasi-triangular formado entre los carbonos C1-C4-C3, y transposición del hidruro H11 desde el átomo C1 hacia el átomo C4.
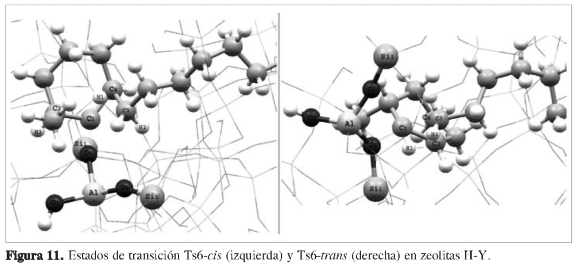
La etapa final de la reacción corresponde nuevamente a la transposición de la cadena alifática desde el carbono C4 hacia el carbono C1, con ruptura de los enlaces C4-C3 y C1-O1, y la formación de un nuevo enlace C3-C1. Simultáneamente se transpone el hidruro H11 desde el átomo C1 hacia el átomo C4, obteniéndose un alcóxido terciario. Esto ocurre mediante estados de transición bicíclicos cuasi-triangular es (Ts6-cis y Ts6-trans) entre los átomos C1-C4-C3 (Figura 11), lo cual permite obtener los productos finales Int7-(cis,trans) cuyas energías relativas fueron muy cercanas entre sí.
Al final del avance de la reacción, la cadena alifática del hidrocarburo se orienta en los diferentes canales que forman la supercavidad, permitiendo la disminución de las interacciones de repulsión del sistema zeolita-huésped, que favorece la formación de los intermedios finales. En este caso se observó que en el producto Int7-trans, una parte de la cadena se dirige hacia el canal superior de la zeolita permitiendo obtener una energía relativa menor que el intermedio Int1. En el caso del Int7-cis no se observó este fenómeno, pero su energía fue relativamente cercana a la del producto Int7-trans, debido a la orientación del grupo metilo que permitió formar el enlace C-O correspondiente al alcóxido.
Si bien las energías relativas de los productos Int7-cis e Int7-trans son cercanas, hay que resaltar que durante el avance de la reacción la ruta a través de aductos trans presentó una caída significativa de la energía relativa en la etapa Int4-trans. Esto implica que el producto que se formará mayoritariamente es el 1-heptil-trans-4-metilciclohexano, que coincide con los datos reportados en la literatura (7, 8). Sin embargo, la ruta cis indica que también es posible obtener el isómero 1-heptil-cis-4-metilciclohexano.
Por otro lado, la energía relativa para los intermedios Int8-(cis,trans) es menor en comparación con la especie de partida, siendo -5,32 kJ/mol para cis y -18,09 kJ/mol para el aducto trans. Esto sugiere la posibilidad de obtener un equilibrio con los intermedios Int7-(cis,trans), aunque necesariamente se debería localizar un posible estado de transición en esta etapa de la coordenada de reacción.
Para la configuración B, las energías relativas para los intermedios y estados de transición fueron representadas mediante un perfil de energía, como lo muestra la Figura 12. La configuración B es descrita de manera similar en la configuración A en cuanto a la forma de cada uno de los intermedios y estados de transición.
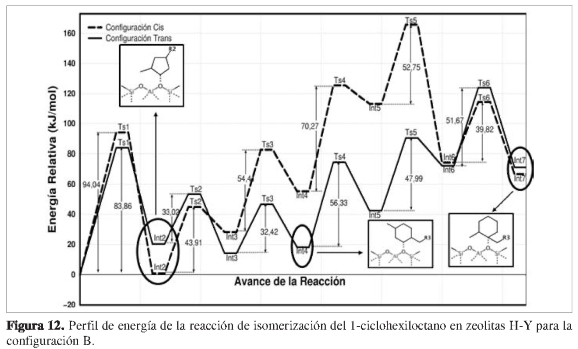
Sin embargo, se diferenció por la disposición del oxígeno ubicado en la boca del poro de la zeolita y por la manera como se orientaba el hidrocarburo para formar cada uno de los intermedios de la reacción, permitiendo obtener energías relativas totalmente diferentes a las calculadas en la configuración A.
A medida que avanzó la reacción, las energías relativas de los aductos cis y trans en cada intermedio aumentaron drásticamente. Es el caso de los intermedios Int5-(cis,trans) e Int7-(cis,trans). En los intermedios Int5-cis e Int5-trans, ocurre la formación de alcóxidos terciarios debido a la transferencia previa de la cadena alifática hacia el átomo C1, originando un fuerte impedimento estérico para estabilizar el carbono electrodeficiente. Finalmente, la reacción de isome-rización termina con la formación de los productos Int7-cis e Int7-trans, cuyas energías relativas fueron 66,65 kJ/mol y 71,13 kJ/mol, respectivamente.
Por último se observó que los intermedios Int8-(cis,trans) poseen energías relativas de 65,30 kJ/mol para cis y 67,80 kJ/mol para trans, revelando el aumento progresivo de la energía durante el avance de la reacción.
CONCLUSIONES
El perfil de energía correspondiente a la isomerización del 1-ciclohexiloctano en fase gaseosa mostró que no se presenta la formación preferencial de aductos cis o trans, debido a que la cadena alifática no aporta densidad electrónica para estabilizar energéticamente los estados de transiciónparaaductos cis ytrans. Sinembargo, seestablecióqueenlacoordenadadereac-ción en fase gaseosa se obtienen preferencialmente carbocationes terciarios caracterizados por la cercanía entre los grupos n-octil y metil, lo cual estabiliza la deficiencia electrónica del átomo carbono en cada intermedio siguiendo el orden de estabilidad característico de los iones carbenios (terciario>secundario>primario).
Los perfiles de energías correspondientes a la isomerización del 1-ciclohexiloctano, catalizadas por zeolitas H-Y y H-ZSM-22, indicaron que se obtiene preferencialmente el isómero 1-heptil-4-trans-metilciclohexano a través de la configuración A, debido a la facilidad para estabilizar el carbono electrodeficiente de la cadena hidrocarbonada a través de enlaces alcóxido con los sitios activos de cada zeolita. Sin embargo, la reacción de isomerización en la zeolita H-ZSM-22 mostró que la energía relativa de intermedios, cis y trans, fueron mayores que el alcóxido Int1 debido a la dificultad en el reordenamiento de la cadena hidrocarbonada dentro de los canales unidimensionales del material para estabilizar el enlace alcóxido.
En la zeolita H-Y, únicamente la configuración A permitió obtener intermedios altamente estables tales como el Int2-cis, Int3-(cis,trans), Int4-trans, Int6-(cis,trans) e Int7-(cis,tras), debido a la libertad que tuvo la cadena alifática para orientarse hacia los diferentes canales que se interceptan para formar la supercavidad. En estas condiciones, se favoreció la formación de isómeros ciclopentil- y ciclohexil-dialquil sustituidos. Las reacciones de isomerización de hidrocarburos catalizadas por zeolitas bifuncionales dependen de la disponibilidad espacial de los oxígenos de la zeolita, del diámetro y morfología de los canales de las zeolitas, de la forma del hidrocarburo y de cada uno de los intermedios y estados de transición generados en el transcurso de la reacción.
AGRADECIMffiNTOS
Los autores agradecen a Colciencias por el apoyo financiero (Proyecto: 110205-16922).
REFERENCIAS BIBLIOGRÁFICAS
1. Despres, J.; Koebel, M.; Krocher, O.; Elsener, M.; Wokaun, A. Adsorption and desorption of NO and NO2 On Cu-ZSM-5. Micropor. Mesopor. Mater. 2003. 58(2): 175-183. [ Links ]
2. Zhu, L.; Zhu; Seff, K. Rate of Tl + exchange into single crystals of Zeolite X. Micropor. Mesopor. Mater. 2000. 39(1-2): 187-193. [ Links ]
3. Thybaut, J. W.; Marin, G. B.; Baron, G. V.; Jacobs, P. A.; Martens, J. A. alkene protonation enthalpy determination from fundamental kinetic modeling of alkane hydroconversion on Pt/H-(US)Y-Zeolite. J. of Cat. 2002. 202(2): 324-339. [ Links ]
4. Maesen, Th. L. M.; Schenk, M.; Vlugt, T. J. H.; Smit, B. Differences Between MFI and MEL-Type Zeolites in paraffin hydrocracking. J. of Cat. 2001. 203(2): 281-291. [ Links ]
5. Coonradt, H. L.; Garwood, W. E. Mechanism of hydrocracking. Reactions of Paraffins and Olefins. Ind. Eng. Chem. Process Des. Dev. 1964. 3(1): 38-45. [ Links ]
6. Ocakoglu, R. A.; Denayer, J. F. M.; Marin, G. B.; Martens, J. A.; Baron, G. V. Tracer chromatographic study of pore and pore mouth adsorption of linear and monobranched al-kanes on ZSM-22 Zeolite. J. Phys. Chem. B. 2003. 107(1) 398-406. [ Links ]
7. Souverijns, W.; Houvenaghel, A.; Feijen, E. J. P.; Martens, J. A.; Jacobs, P. A. Isomerization of 1-Cyclohexyloctane on Pt/H-ZSM-22 bifunctional zeolite catalyst. J. of Cat. 1998. 174(2): 201-209. [ Links ]
8. Souverijns, W.; Parton, R.; Martens, J. A.; Froment, G. F.; Jacobs, P. A. Mechanism of the paring reaction of naphtenes. Cat. Lett. 1996. 37(3-4): 207-212. [ Links ]
9. Chica, G. A.; Corma, A. On the mechanism of alkane isomerisation (Isodewaxing) with unidirectional 10-Member ring zeolites. A molecular dynamics and catalytic study. J. of Cat. 2000. 195(2): 227-236. [ Links ]
10. Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993. 98: 5648-5652. [ Links ]
11. Ditchfield, R.; Hehre, W. J.; Pople, J. A. Self-Consistent molecular-Orbital Methods. IX. An extended gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971. 54: 724-728. [ Links ]
12. Halgren, T. A.; Lipscomb, W. N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977. 49(2): 225-232. [ Links ]
13. González, C.; Schlegel, H. B. An improved algorithm for reaction path following. J. Chem. Phys. 1989. 90: 2154-2161. [ Links ]
14. Marles, B. Silica-ZSM-22: Synthesis and single crystal structure refinement. Zeolites. 1987. 7(5): 393-397. [ Links ]
15. Jirák, Z.; Vratislav, S.; Bosácek, V. A neutron diffraction study of H-, Na-Y zeolites. J. Phys. Chem. Sol. 1980. 41(10): 1089-1095. [ Links ]
16. Kazanksy, V. B.; Senchenya, I. N. Quantum Chemical study of the electronic structure and geometry of surface alkoxy groups as probable active intermediates of heterogeneous acidic catalysts: what are the adsorbed carbenium ions? J. of Cat. 1989. 119(1): 108-120. [ Links ]
17. Rigby, A. M.; Kramer, G. J.; Van Santen, R. A. Mechanisms of hydrocarbon conversion in zeolites: A Quantum mechanical study. J. of Cat. 1997. 170(1): 1-10. [ Links ]
18. Maseras, F.; Morokuma, K. IMOMM: A new integrated Ab Initio+Molecular mechanics geometry optimization scheme of equilibrium structures and transition states. J. Comp. Chem. 1995. 16(9): 1170-1179. [ Links ]
19. Rappe, A. K.; Casewit, C. J.; Colwell, K. S.; Goddard, W. A.; Skiff, W. M. UFF, A full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992. 114(25): 10024-10035. [ Links ]
20. Gaussian 03, Revision D.01. Wallingford CT: Gaussian, Inc. 2004. [ Links ]
21. Peng, C.; Ayala, P. Y.; Schlegel, H. B.; Frisch, M. J. Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comp. Chem. 1996. 17(1): 49-56. [ Links ]

