










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804On-line version ISSN 2357-3791
Rev.Colomb.Quim. vol.38 no.3 Bogotá Sep./Dec. 2009
ESTUDIO TEÓRICO DEL VIRUS DEL SÍNDROME RESPIRATORIO AGUDO SEVERO (SARS) A TRAVÉS DEL USO DE MÉTODOS DE ACOPLAMIENTO MOLECULAR
THEORETICAL STUDIES OF THE VIRUS SEVERE ACUTE RESPIRATORY SYNDROME (SARS) THROUGHT OF THE USE OF METHODS OF MOLECULAR DOCKING
ESTUDO TEÓRICO DO VÍRUS DA SÍNDROME RESPIRATÓRIA AGUDA SEVERA (SARS) A TRAVES DO USO DE MÉTODOS DE ACOPLAMENTO MOLECULAR
Ricardo Vivas-Reyes1, José Luis Ruiz, Roger Caraballo
1 Programa de Química, Facultad de Ciencias Exactas y Naturales, Universidad de Cartagena. Campus de Zaragocilla, Cartagena, Colombia. rvivasr@unicartagena.edu.co
Recibido: 14/07/09 - Aceptado: 26/11/09
RESUMEN
En este estudio se ha evaluado por medio de la metodología de acoplamiento molecular una serie de 5 ligandos borados, que son variantes de la molécula FL-078. Estas moléculas tienen actividad inhibitoria frente a la proteasa Mpro responsable de la replicación del SARS-CoV.
Haciendo uso del programa SYBYL7.0 se optimizó el homodímero de la Mpro (código 1Q2W), y a través del software FlexX se hizo el acople molecular con el fin de escoger el confórmero más estable de los ligandos aril borados frente a la macromolécula Mpro, encontrándose que la estructura FL-166 fue la de mejor conformación.
Los 5 ligandos aril borados tienen la propiedad de interaccionar con el grupo hidroxilo (OH) presente en los residuos tales como serinas, treoninas y tirosinas. Los resultados acople molecular muestran que el mejor acercamiento sobre la cavidad se da sobre el conjunto de treoninas 21, 24, 25 y 26, y no como se afirma en la literatura: que se da sobre el conjunto de serinas 139, 144 y 147.
Palabras clave: síndrome respiratorio agudo severo, acoplamiento molecular, FlexX, inhibición.
ABSTRACT
In this study a set of 5 aryl boronic ligands which are variations of the FL-078 molecule were assessed. These molecules haveinhibitoryactivityagainstoftheMpro protease involved in the SARS-CoV replication.
A homodimeroftheMpro(ID1Q2W) was optimized using the SYBYL7.0 package and through the FlexX software was calculated the molecular docking withthe aim of choose the most stable ligand front to Mpro macromolecule. Was found among the five boronic ligands that the best pose corresponded to the FL-166 structure.
These molecules show interaction with the hydroxil group beared by aminoacid residues such as serines, threoni-nes and tyrosines. The docking calculation results presented here show that the best approaching over the cavity occur on the threonines set 21, 24, 25 and 26 instead of the set pointed out by other authors Serines 139, 144 and 147.
Key words: Severe acute respiratory syndrome, docking molecular, FlexX , inhibition.
RESUMO
Neste estudo avaliou-se através da metodologia do acoplamento molecular uma serie de 5 ligandos contendo boro, que são variantes da molécula FL-078, estas moléculas têm atividade inibitória à protease Mpro responsável da replicação do SARS-CoV.
Usando o programa SYBYL7.0 se otimizou o homodímero da Mpro (código 1Q2W), e através do software FlexX se realizou o acoplamento molecular com a finalidade de escolher o confórmero mais estável dos ligandos aril borados frente à macromolécula Mpro. Encontrando-se que a estrutura FL-166 foi a de melhor conformação.
Os 5 ligandos aril borados têm a propriedade de interagir com o grupo hidro-xila (OH) presente nos resíduos tais como serinas, treoninas e tirosinas. Os resultados de acoplamento molecular mostraram que a melhor aproximação sobre a cavidade ocorre sobre o conjunto de treoninas 21, 24, 25 y 26 e não como se afirma na literatura que se da sobre o conjunto de serinas 139, 144 y 147.
Palavras-chave: Síndrome respiratória aguda severa, acoplamento molecular, FlexX, inibição.
INTRODUCCIÓN
En los primeros años del presente siglo, la aparición del virus del Síndrome Respiratorio Agudo Severo (SARS) (1), y su rápida expansión, puso a muchos científicos y autoridades de salud a nivel mundial a pensar en cómo desarrollar potentes fármacos que fuesen eficaces en la inhibición del virus y, por tanto, en su replicación (2). En general esta clase de estudios debe partir del conocimiento bioquímico, morfológico y de los mecanismos del funcionamiento del virus.
La principal proteinasa del coranovirus del SARS es la 3CLpro oMpro (3), que desempeña un papel importante en la replicación del virus y está formada por dos subunidades que tienen un sitio activo en el cual se encuentra la díada catalítica formada por la histidina-41 y cisteína-145 (4-7). Debido a que la Mpro es fundamental en la replicación del virus, se considera como un blanco relevante para el diseño de drogas y de posibles inhibidores en contra del virus del SARS.
El diseño de posibles inhibidores para la Mpro del SARS-CoV es posible hoy en día gracias al uso de herramientas bioinformáticas. Entre las herramientas bioinformáticas más usadas para el diseño molecular está la técnica del acoplamiento molecular (8, 9). Esta técnica permite la generación de múltiples confórmeros para cada ligando, que son probados con diferentes ligandos sobre diferentes sitios de una proteína dada (10). Dependiendo del software utilizado, diferentes búsquedas conformacionales se pueden dar dependiendo del método utilizado (11-17). Independientemente del método aplicado, el proceso de encontrar confórmeros óptimos debe ser guiado por la función de evaluación, la cual se usa para evaluar las diferentes interacciones fisicoquímicas entre la molécula del ligando y la proteína. En este estudio se utilizó el programa FlexX (8) para la evaluación de cinco ligandos del tipo aril bóricos que poseen actividad inhibitoria; estas 5 estructuras moleculares presentan actividad inhibitoria, la cual ha sido evaluada experimentalmente (18).
MATERIALES Y MÉTODOS
La estructura tridimensional (3D) de la molécula Mpro fue obtenida a través del Protein Data Bank (PDB), bajo el código ID 1Q2W (19); esta molécula es un ho-modímero. La modelización, visualiza-ción y optimización de esta molécula se realizaron con la ayuda del paquete de programas computacionales Sybyl (20). Los átomos de hidrógeno y pares solitarios fueron agregados a la estructura cristalina de rayos X usando el módulo Biopolimer de Sybyl. La orientación inicial de los hidrógenos fue la misma utilizada en el diccionario de definiciones del programa (20). Hay que aclarar en este punto que todos los cálculos teóricos fueron hechos para un monómero, porque se infiere que el otro monómero actúa de manera similar por tener igual estructura y secuencia peptídica. La optimización del monómero se realizó usando el campo de fuerza MMFF94s (21), con un criterio de convergencia para el gradiente de energía de 0,05 kcal mol-1 usando el método del gradiente conjugado (22). La estructura de los ligandos (Figura 1) fue optimizada usando el programa Gaussian (23) a un nivel de cálculo AM1 (24).
Acoplamiento molecular
Los cálculos de acoplamiento molecular de la Mpro con los compuestos bóricos se implementaron usando el módulo FlexX (8), incluido en el paquete computacional de SYBYL 7.0 (20). De este paquete computacional se empleó el módulo Cscore como función de evaluación de los resultados (25, 26) arrojando alrededor de 100 conformaciones por ligando en el sitio activo, las cuales fueron definidas como todos los residuos encontrados dentro de un radio de 6,5 Á de la Cys-145 e His-41 de la proteasa Mpro del virus del SARS. El programa FlexX automáticamente coloca los ligandos dentro del sitio activo previamente definido, considerando las restricciones tanto de tipo geométrico como electrónico.
Consenso molecular
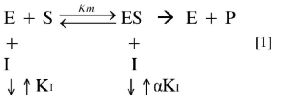
Se eligió la conformación molecular más favorable de la serie de ligandos después de hacerle su respectivo acoplamiento molecular; igualmente se realizó una comparación entre los diferentes parámetros de valoración, a través de un consenso que se desarrolla entre las funciones de evaluación. Las funciones de evaluación más frecuentemente utilizadas son: Chemscore (27), D-score (28), PMF-score (29) and Gscore (13) y Total-Score de FlexX (8). La función Cscore categoriza las energías en orden de 0 a 5 las mejores energías acomplejantes. Sin embargo, cada función de evaluación selecciona también su mejor conformación y es por tal motivo que estas se clasifican en el orden de mayor a menor energía como números enteros de menor a mayor, respectivamente, y así, después de realizar la media, el que tenga número entero de menor valor será el que tenga el valor energético mayor.
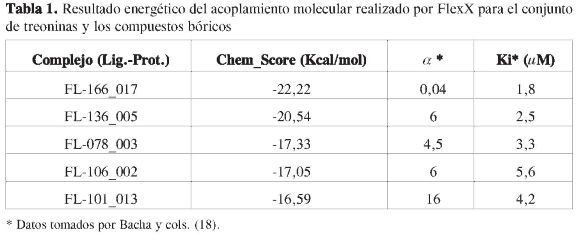
Después de elaborar el consenso, se tomó el valor ChemScore como referencia para compararlo con los resultados experimentales (21). Estos valores son mostrados en la Tabla 1, en donde los valores de la constante de inhibición (Ki) y el parámetro a indican la competitividad de los ligandos, es decir, que entre más bajo sea el valor de aKi, la inhibición será entonces no competitiva.
Datos moleculares
Se ha reportado en la literatura una serie de compuestos borados (18), (Figura 1), que experimentalmente presentan actividad antiviral frente a la proteasa Mpro mostrando selectividad hacia los residuos de aminoácidos que poseen grupos OH, tales como treoninas y serinas. Los compuestos aril bóricos en general han sido usados en diversas áreas de la química, tales como química organometálica, síntesis orgánica y química medicinal (30). Por ejemplo los ácidos fenil bóricos (31, 32), como también algunos otros aril bóricos (33) han demostrado tener actividad inhibitoria débil para la serina proteasa. La inhibición más potente de la serina proteasa se ha observado para los ácidos bóricos incorporados dentro de un esqueleto péptido mimético (34, 35). En la actualidad existen reportes de una gama de inhibidores peptidilborano (36, 37).
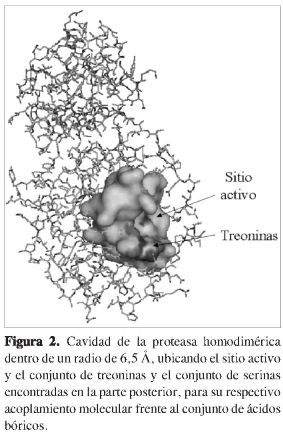
El sitio activo de la estructura de la Mpro está constituido por histidina 41-cisteína 145 (14). Se eligió una distancia de 6,5 Å alrededor de la cisteína 145; en este radio existen unos conjuntos bien conformados de treoninas [21, 24, 25 y 26] y serinas [139, 144, 147], que forman una cavidad que resultó ser del tamaño adecuado para retener a los compuestos aril bóricos.
RESULTADOS Y DISCUSIÓN
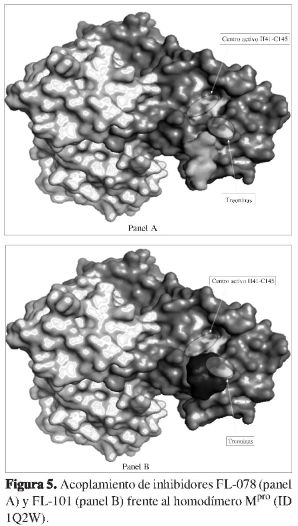
A partir de los cálculos de acoplamiento molecular hechos sobre la cavidad de las serinas [139, 144, 147] (Figura 2), se encontró que los resultados sobre esta cavidad difieren de los reportados en la literatura (18), en los que se afirma que la inhibición se desarrolla debido al hecho de que los compuestos aril bóricos (Figura 1) interaccionan con los hidroxilo (OH) de las serinas, lo cual propicia un acercamiento hacia la díada catalítica conformada por la histidina y cisteína [His 41-Cys 145] (4-7). Sin embargo, a través de este estudio se demuestra claramente que el acercamiento realmente no es sobre la cavidad rica en serinas, sino que se da sobre la cavidad que posee el grupo hidroxilo (OH) en su estructura química, y que está conformado por un conjunto de treoninas 21, 24, 25 y 26, difiriendo en este caso con lo reportado por Bacha y colaboradores (18) como se puede observar en las Figuras 3 y 4.
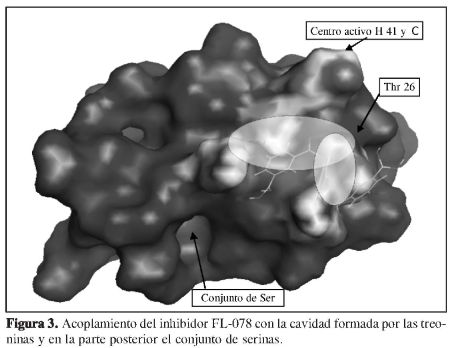
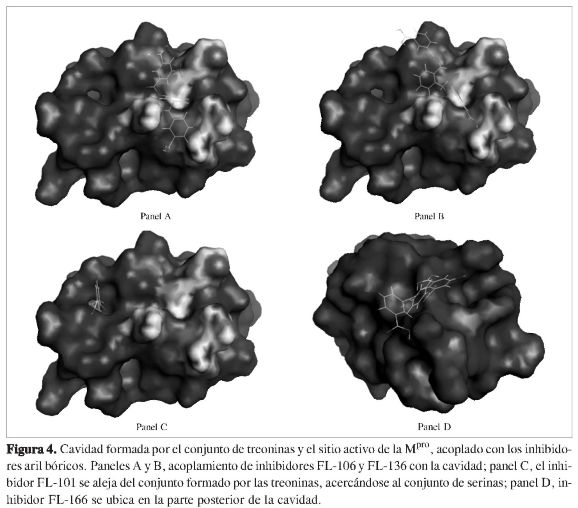
El acoplamiento realizado sobre la Mpro con los diferentes ligandos bóricos muestra que estos pueden ser posicionados adecuadamente en algún sitio activo de una proteína, pero la fortaleza de las interacciones no son suficientemente grandes dentro del complejo enzima-ligando como para mantenerlo unido de forma estable, lo cual hace que este ligando no pueda competir contra el sustrato asociado a la proteína.
En la Tabla 1 se muestran los valores energéticos del acople molecular realizado sobre la estructura de la Mpro con los li-gandos bóricos; estos valores indican que a pesar de la posición en la que está ubicado el ligando bórico dentro de la estructura acomplejante FL-101013, con un orden de energía de -16,59 Kcal/mol, a partir de este valor energético puede asumirse que el complejo presentará interacciones débiles si se compara con los otros casos, y por tanto el complejo enzima-inhibidor (FL-101013) será el más inestable, como también se puede inferir de su constante de inhibición 16,0,µM, el parámetro a = 4,2 eleva el valor de la constante a aKi = 67,2,µM, (4), lo cual corrobora que a pesar de que su estructura, como se muestra en la Figura 5 (panel B), se encuentra cercano a la díada histidina 41 y cisteína 145, la fortaleza de las interacciones no es suficientemente grande como para mantenerlo unido en forma estable.
Análogamente se tienen las estructuras FL-136 y FL-106; estas moléculas tienen una constante de inhibición de 6,0 ,µM, que experimentalmente las clasifica como mejores que la estructura FL-101. Al analizar esto desde el punto de vista de la superficie molecular formada entre el ligando con el conjunto de treoninas, se tiene que la FL-106, con una energía de -17,05 Kcal/mol, hace que las interacciones energéticas sean por -0,46 Kcal/mol más estables que la FL-101, aunque tanto la FL-106 como la FL-136 se ubican dentro de la misma zona lejana de la histidina 41 y cisteína 145 debido a las constantes inhibitorias y los valores de las interacciones que presentan -17,05 y -20,54 Kcal/mol, respectivamente. Como se observa en la Tabla 1, estos ligandos tienen el mismo valor de la constante de inhibición (18), se diferencian en los valores que se encuentran en el producto de esta constante y el parámetro de inhibición aKi, los cuales son, respectivamente, 15,00 µM y 33,60 µM para la FL-136 y la FL-106.
La constante de inhibición del complejo formado entre la proteasa y el ligando FL-078, 4,5 µM, está dentro de los valores hallados anteriores (de 6 µM) ya que energéticamente posee interacciones que caen dentro del rango anterior (-17,05 y -20,54 Kcal/mol) y su valor energético (-17,33 Kcal/mol), y posible que este ligando no siga la tendencia experimental debido quizás a que no se tuvo en cuenta el efecto que puede generar el solvente en la estructura proteínica, el efecto salino o de iones y el pH, entre otros; pero es bastante claro que al observar las estructuras de la Figura 6, correspondientes a la de las FL-078, FL-106 y FL-136, respectivamente, se puede apreciar que las tres estructuras están en la misma zona, y estructuralmente es muy probable que esto se refleje en sus actividades cercanas, ya que, como se ha planteado, puede tratarse de un sitio con posible actividad.
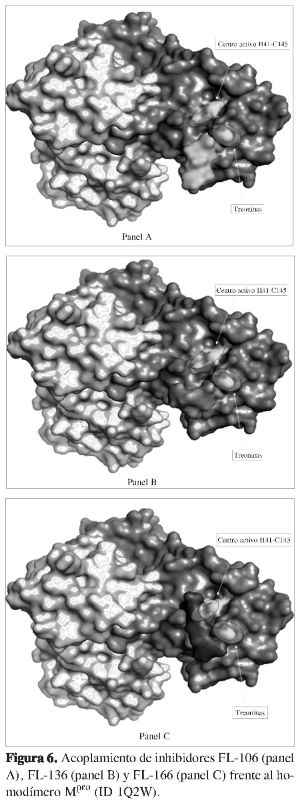
Por último, se tiene la estructura del ligando FL-166 (Figura 6, panel C); se acompleja con la Mpro con una energía de -22,22 Kcal/mol y con un valor de 0,04 µM en su constante de inhibición. Estos datos muestran que esta es la mejor estructura, estando este resultado con lo planteado por Bacha (18), debido al intercambio de grupos ester por grupos amidas, lo cual proporciona interacciones mucho más fuertes desde el punto de vista energético, a diferencia de todas las demás. No obstante, la ubicación molecular de este ligando está hacia el centro de la díada catalítica (14), pero puede decirse que es estabilizado debido a la interacción que ocurre con el residuo Thr 25, y muy especialmente con el residuo Asn 119, los cuales se ubican dentro de la zona que se ha mencionado repetidamente para las estructuras anteriores como un posible sitio donde los ácidos bóricos presentan actividad; sin embargo, el valor para el parámetro de inhibición es aKi 0,078 y la clasifica como una estructura que por su ubicación en el centro activo histidina 41 cisteína 145 (14) puede interferir competitivamente con el sustrato y con la díada catalítica.
La tendencia energética calculada por el acoplamiento molecular de la Mpro ylos complejos es FL-166_017 > FL-136_005 > FL-078_003 > FL-106_002 > FL-101_013; al compararla con el comportamiento dado por el producto del parámetro alfa y la constante de inhibición (Tabla 1) indican que el parámetro alfa es el que tiene el mayor peso sobre la tendencia para el mecanismo de inhibición no competitiva, como se observa en la ecuación 1 y la Figura 7:
CONCLUSIONES
A partir de lo expuesto anteriormente se puede concluir que los compuestos bifuncionales, tales como los aril bóricos y sus diversas variaciones, tienen gran selectividad por el grupo hidroxilo (OH) que se encuentra en el residuo de la treonina, que es un aminoácido pequeño, polar y que presenta en su estructura molecular un alcohol primario en su cadena lateral, que puede actuar como nucleófilo durante las reacciones de catálisis enzimática. El compuesto aril bórico codificado como FL-166 presenta una alta inhibición enzimática, lo cual se evidenció a través de su energía de interacción proteína-Ligando -22,22 Kcal/mol y por su constante de inhibición 0,04 µM determinada experimentalmente. Este compuesto podría ser también blanco de futuras variaciones estructurales.
La estructura del ligando FL-166 acomplejada con la Mpro es la mejor estructura frente a los cinco compuestos reportados por Bacha, debido al intercambio de grupos ester por grupos amidas, lo cual proporciona interacciones mucho más fuertes desde el punto de vista energético, a diferencia de todas las demás. No obstante, la ubicación molecular de este ligando está dirigida hacia el centro de la diada catalítica, pero puede decirse que es estabilizado debido a la interacción que ocurre con el residuo Thr 25, y muy especialmente con el residuo Asn 119, los cuales se ubican dentro de la zona que se ha mencionado repetidamente para las estructuras anteriores como un posible sitio donde los ácidos bóricos presentan actividad, sin embargo, este valor para el parámetro de inhibición aKi 0,078 obtenido experimentalmente, la clasifica como una estructura que por su ubicación en el centro activo histidina 41, cisteína 145, puede interferir competitivamente con el sustrato y con la díada catalítica.
El comportamiento dado por el producto de aKi indica que a es la que marca la tendencia para el mecanismo de inhibición no competitiva al compararlo con los resultados energéticos dados a través de los cálculos de acople molecular.
AGRADECIMIENTOS
Los autores agradecen a la Universidad de Cartagena y a Colciencias por el soporte económico prestado para el desarrollo de este trabajo, en especial al Programa de Jóvenes Investigadores financiado por las dos instituciones.
REFERENCIAS BIBLIOGRÁFICAS
1. WHO. Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. World Health Organization, 2003. [ Links ]
2. Lee,N.;Hui,D.; Wu, A.; Chan, P.: Cameron, P.: Joynet, G. M.; Ahuja, A.; Yung, M. Y.; Leung, C. B.; To, K. F.; Lui, S. F.; Szeto, C. C.; Chung, S.; Sung, J. J. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl J Med. 2003. 348 (20): 1986-1994. [ Links ]
3. Anand, K.; Gottfried, J.; Jeroen, R.; Stuart, G.; Ziebuhr, J.; Hilgenfeld, R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra a-helical domain. EMBO J 2002. 2: 3213-3224. [ Links ]
4. Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J. R.; Hilgenfeld, R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science. 2003. 300: 1763-1767. [ Links ]
5. Matthews, D.; Smith, W.; Ferre, R.; Condon, D.; Budahazi, D.; Sisson, W.; Villafranca, J.; Janson, C.; McElroy, H.; Gribskov, C. Structure of human rhinovirus 3C protease reveals a trypsin-like polypeptide fold, RNA-binding site, and means for cleaving precursor polyprotein. Cell, 1994. 77(5): 761-771. [ Links ]
6. Chou, C.-Y.; Chang, H.-C.; Hsu, W.-C.; Lin, T.-Z.; Lin, C.-H.; Chang, G.-C. Quaternary Structure of the Severe Acute Respiratory Syndrome (SARS) Coronavirus Main Protease. Biochemistry. 2004. 43 (47): 14958-14970. [ Links ]
7. Fan, K.; Ma, L.; Han, X.; Liang, H.; Wei, P.; Liu, Y.; Lai, L. Mutational and inhibitive analysis of SARS coronavirus 3C-like protease by fluorescence resonance energy transfer-based assays. Biochem. Biophys. Res. Commun. 2005. 329: 934-940. [ Links ]
8. Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algoritm. J Mol Biol. 1996. 261 (3): 470-489. [ Links ]
9. Stahl, M.; Rarey, M. Detailed Analysis ofEvaluacionFunctionsfor Virtual Screening. J. Med. Chem. 2001. 44: 1035-1042. [ Links ]
10. (a) Kuntz, I. D. Structure-ased strategies for drug design and discovery. Science. 21, 257, 1078-1082. (b) Kunz, I. D.; Meng, E. C.; Schoichet, B. K. Structure-based molecular design. Acc Chem Res. 1994. 27 (5): 117-123. [ Links ]
11. Ian, L. A.; Nikolay, T. P.; Philip, M. D. J. Med. Chem. 2005. 48: 6585-6596. [ Links ]
12. Jones, G.; Willett, P.; Glen, R. C.; Leach, A. R.; Taylor, R. Develop-mentandvalidationofageneticalgo-rithm for flexible docking. J Mol Biol. 1997. 267 (3): 727-748. [ Links ]
13. Morris, G. M.; Goodsell, D. S.; Halliday, R. S.; Huey, R.; Hart, W. E.; Belew, R. K.; Olson, A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998. 19: 1639-1662. [ Links ]
14. Goodsell, D. S.; Olson, A. J. Automated docking of substrates to proteins by simulated annealing Proteins. Struct. Funct. Genet. 1990. 8: 195-202. [ Links ]
15. Trosset, J. Y.; Scheraga, H. A. PRODOCK: software package for protein modeling and docking. J. Comput. Chem. 1999. 20: 412-427. [ Links ]
16. Leach, A. R.; Smellie, A. S. A Combined Model-Building and Distance Geometry Approach to Automated Conformational Analysis and Search? J. Chem. Inf. Comput. Sci. 1992. 32: 379-385. [ Links ]
17. Klebe, G.; Mietzner, T. A fast and efficient method to generate biologically relevant conformations. J. Comput. Aided Mol. Des. 1994. 8: 583-606. [ Links ]
18. Bacha, U.; Barrila, J.; Velázquez-Campoy, A.; Leavitt, S. A.; Freire, E. Identification of Novel Inhibitors of the SARS Coronavirus Main Protease 3CLpro. Biochemistry. 2004. 43 (17): 4906-4912. [ Links ]
19. Bonano, J. B.; Fowler, R.; Gupta, S.; Hendle, J.; Lorimer, D.; Romero, R.; Sauder, M.; Wei, C. L.; Liu, E. T.; Burley, S. K.; Harris, T. X-Ray Crystal Structure of the Sars Coronavirus Main Protease. Available from: http://www.rcsb.org/pdb/cgi/explore.cgi?pdbId=1a2w.(2003). SYBYL 7.3. [ Links ]
20. Tripos International, 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA. URL: http://www.tripos.com/ [ Links ]
21. Halgren, T. A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999. 20 (7): 720-729. [ Links ]
22. Press, W. H.; Flannery, B. P.; Teukolsky, S. A.; Vetterling, W. T. Numerical Recipes in C, The Art of Scientific Computing, Cambridge University Press, 1988. [ Links ]
23. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Pople, J. A. et al. Gaussian 98, Revision A.9, (1988) Gaussian, Inc., Pittsburgh PA. [ Links ]
24. Dewar, M.; Zoebisch, E.; Stewart, J. AM1: A New General Purpose Quantum Mechanical Molecular Model. Amer. Chem. Soc. 1985. 107: 3902-3909. [ Links ]
25. Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical evaluacion functions for structure-based binding affinity prediction. J Comput-Aided Mol Des. 2002. 16 (2): 11-26. [ Links ]
26. Clark, M.; Cramer, R.; Van Opdenbosch, N. Validation of the general purpose tripos 5. 2 force field. J Comput Chem. 1989. 10 (8): 982-1012. [ Links ]
27. Eldridge, M. D.; Murray, C. W.; Aurton, T. R.; Paolini, G. V.; Mee, R. P. Empirican Evaluacion Functions: I. The development of a fast empirical evaluacion function to estimate the binding affinity ofligands in receptor complexes. J Comput -AidedMolDes. 1997. 5: 425-445. [ Links ]
28. Kuntz, J.; Blaney, J. M.; Oatley, S.; Langridge, J. R.; Ferrin, T. E. A. A geometric approach to macromole-cule-ligand interactions J. Mol. Biol. 1982. 161: 269-288. [ Links ]
29. Muegge, I.; Martin, Y. A general and fast evaluacion functions for protein-ligand interactions: a simplified potential approach. J Med Chem. 1999. 42 (5): 791-804. [ Links ]
30. Lazarova, T. I.; Jin, L.; Rynkiewicz, M.; Gorga, J. C.; Bibbins, F.; Meyers, H. V.; Babine, R.: Strickler, J. Synthesis and in vitro biological evaluation of aryl boronic acids as potential inhibitors of factorXIa. Bioorg Med Chem Lett. 2006. 16 (19): 5022-5027. [ Links ]
31. Weston, G. S.; Blazquez, J.; Baquero, F.; Shoichet, B. K. Structure-based enhancement of boronic acid-based inhibitors of AmpC beta-Lactamase. J Med Chem. 1998. 41 (23): 4577-4586. [ Links ]
32. Sieber, J. D.; Morken, J. P. Sequential Pd-catalyzed asymmetric llene diboration/alpha-aminoallylation. J Am ChemSoc. 2006. Jan 11.128 (1): 74-75. [ Links ]
33. Spencer, J.; Burd, A. P.; Goodwin, C. A.; Merette, S. A. M.; Scully, M. F.; Adatia, T. et al. Synthesis of ortho-modified mercapto- and piperazi-no-methyl-phenylboronic acid derivatives. Tetrahedron. 2002. 58 (8): 1551-1556. [ Links ]
34. Bukhtiyarova, M.; Rizzo, C. J.; Kettner, C. A.; Korant, B. D.; Scarnati, H. T.; King, R. W. Inhibition of the bovine viral diarrhoea virus NS3 serine protease by a boron-modified peptidyl mimetic of its natural substrate. Antiviral chem chemother. 2001. 6: 367-373. [ Links ]
35. Jagannathan, S.; Forsyth, T. P.; Kettner, C. A. Synthesis of boronic acid analogues of alpha-amino acids by introducing side chains as electrophiles. J Org Chem. 2001. 66 (19): 6375-6380. [ Links ]
36. Coburn, C. A. Small-molecule direct thrombininhibitors: 1997-2000. Expert Opin Ther Patents. 200 (5): 721-738. [ Links ]
37. Ni, W.; Kaur, G.; Sprimgsteen, G.; Wang, B.; Franzen, S. Regulating the fluorescence intensity of an anthracene boronic acid system: a B-N bond or a hydrolysis mechanism? Bioorg Chem. 2004 . 32 (6): 571-581. [ Links ]

