










Serviços Personalizados
Journal
Artigo
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Colombiana de Química
versão impressa ISSN 0120-2804versão On-line ISSN 2357-3791
Rev.Colomb.Quim. v.39 n.1 Bogotá jan./abr. 2010
QUANTITATIVE STRUCTURE-ACTIVITY RELATIONSHIPS TO PREDICT IN VITRO TEAC AND EC50 OF SYNTHETIC ANILINES
RELACIONES CUANTITATIVAS ACTIVIDAD-ESTRUCTURA PARA PREDECIR LA TEAC Y LA EC50 DE ANILINAS SINTÉTICAS
RELAÇÕES QUANTITATIVAS ATIVIDADE - ESTRUTURA PARA A PREDIÇÃO A TEACEAEC50 DA ANILINAS SINTÉTICAS
Geovanna Tafurt-García1,2, Jairo René Martínez1, Elena Stashenko1, Leonor Y. Vargas3
1 Escuela de Química, Facultad de Ciencias, Universidad Industrial de Santander. A. A. 678, Bucaramanga, Colombia.
3 Facultad de Química Ambiental. Universidad Santo Tomás. A. A. 1076, Bucaramanga, Colombia.
Recibido: 26/03/09 - Aceptado: 06/04/10
ABSTRACT
Quantitative Structure-Activity Relationships (QSAR) are useful in understanding how chemical structure relates to the biological activity of natural or synthetic compounds and for designing newer and better compounds. In the present study, 22 N-arylmethyl substituted anilines were treated with ABTS (2,2'-azinobis- (3-ethylbenzothiazoline-6-sulfonic-acid)) and DPPH (2,2-diphenyl-1-picrylhy-dracyl) radicals in order to evaluate their TEAC (mmol trolox/mmol antioxidant, Trolox Equivalent Antioxidant Capacity) and EC50 (mmol antioxidant/mmol initial DPPH, Antioxidant Equivalent Concentration to decrease the initial DPPH concentration by 50 %) values, respectively. Different QSARs were developed based on these data, using theoretical descriptors derived from geometry-optimized molecular structures. A model with electronic energy (EE), total charge weighted partial positively charged surface area (PPSA-2), and exact polarizability (αzz) as descriptors showed satisfactory predictive TEAC performance according to internal and external validation procedures. It can be useful in predicting data and setting a testing priority for those compounds not yet synthesized or for which experimental data are not available.
Key words: QSAR, TEAC, EC50,antioxidant capacity, anilines.
RESUMEN
Las relaciones cuantitativas actividad-estructura (QSAR) son útiles para entender la forma en que la estructura química de sustancias sintéticas y naturales se relaciona con la actividad biológica, y para el diseño de nuevos y mejores compuestos. En el presente estudio fueron evaluadas las capacidades de 22 anilinas N-arylmetil sustituidas para la captura de los radicales ABTS (ácido 2,2'-azino-bis(3-etilbenzo-tiazolino-6-sulfónico) y DPPH (2,2-difenil-1-picrilhidracilo), relacionadas con los valores de TEAC (mmol trolox/mmol antioxidante, capacidad antioxidante equivalente al Trolox) y EC50 (concentración equivalente de antioxidante para disminui r la concentración inicial de DPPH enun 50%), respectivamente. Las TEAC, las EC50 y los descriptores teóricos derivados de las estructuras moleculares optimizadas fueron utilizados para elaborar las diferentes QSAR. Los modelos TEAC con descriptores como EE (energía electrónica), PPSA-2 (carga total pesada con el área superficial cargada positivamente) y αzz (polarizabilidad exacta) mostraron una capacidad de predicción satisfactoria por procedimientos de validación interna y externa, por lo que pueden ser útiles para la predicción de actividades de compuestos que aún no han sido sintetizados o con datos experimentales no disponibles.
Palabras clave: QSAR, TEAC, EC50, capacidad antioxidante, anilinas.
RESUMO
Relaçãos quantitativa atividade - estrutura tem sido empregado para estabelecer se a estrutura química está relacionada com a atividade biológica dos químicos naturais e sintéticos e para o desenho de novos e melhores compostos. No presente estudo foram avaliadas as capacidades de 22 anilinas N-arylmetil sustituidas para a captura dos radicais de ABTS (ácido-2,2'-azino-bis(3-etilo-benzo-tiazolino-6-sulfônic o) e DPPH (2,2-difenilo-1-picrilhidracilo), relacionadas com os valores de TEAC (mmol trolox/mmol antioxidante, capacidade antioxidante equivalente ao Trolox) yEC50 (concentração equivalente de antioxidante para a diminuição da concentração inicial de DPPH para um 50 %) respectivamente. As TEAC, as EC50 eos descritores teóricos derivados das estruturas moleculares otimizadas foram empregados para elaborar as diferentes QSARs. Os modelos TEAC com descritores como EE (Energia Eletrônica), PPSA-2 (carga total pesada com a área superficial carregada positivamente) y αzz (polarizabilidade exata) mostraram uma satisfatória capacidade de predição pelos procedimentos de validação interna e externa, e por tanto podem ser úteis para a predição da capacidade antioxidante de compostos que ainda no tem sido sintetizados com dados experimentais não disponíveis.
Palavras-chave: QSARs, TEAC, EC50, Capacidade Antioxidante, Anilines.
INTRODUCTION
Aromatic amines are used as antioxidants in the production of rubber or cutting oils, and in the production of polymers, dyes, pesticides and pharmaceuticals (1). Diarylamines are used as antioxidants in lubricating oils, whereas the benzylami-nes are used as antioxidants and therapeutic agents for disorders of the central nervous system (2, 3).
QSARs have been used for screening chemical databases before the synthesis of chemicals, and for estimating and predicting the biological activity of chemicals (3-7). Several SAR and QSAR have been designed for antioxidant capacity of gallic acids, phenols, coumarins and flavonoids in chemical, enzymatic, and cellular systems (8-13), but only a few QSAR have been developed for benzyla-mine and aniline antioxidants (3-14).
TEAC and EC50 test systems provide reproducible and quantitative results of reactivity to radicals (15-17). They can be used in the development of a model for estimating antioxidant capacity. The aim of this study was to develop multilinear regression (MLR) models to predict reliable TEAC and EC50 values of new anilines with chemical domain similar to the training set. The model predictive capability was statistically evaluated both internally and externally.
MATERIALS AND METHODS
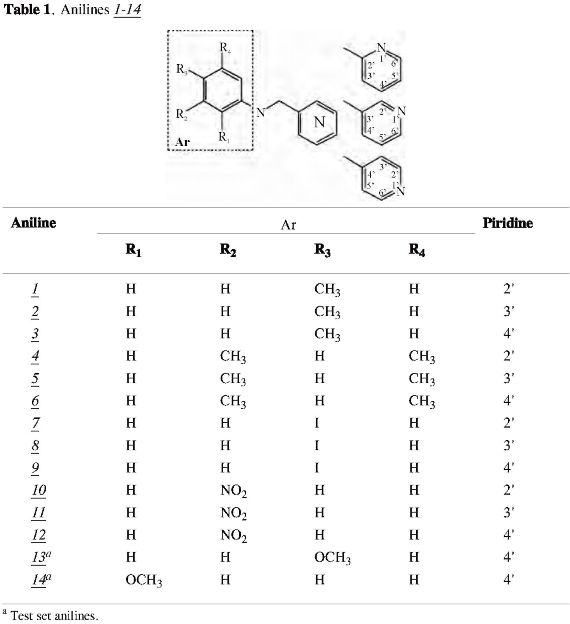
Reactivities of the synthetic anilines to ABTS and DPPH radicals were assessed according to the procedures of R. Re, et al. (15), and W. Brand-Williams et al. (16). DPPH and ABTS were obtained from Sigma-Aldrich Chemie Gmbh (D-9132 and A1888) and methanol and et-hanol, HPLC grade, were purchased from Mallinckrodt (NJ08865). Anilines were prepared in the Laboratory of Organic and Biomolecular Chemistry of the Industrial University of Santander according to the technique described by Kousnetsov et al. (18). The spectrophotometric data were acquired using a JENWAY 6300 spectrophotometer. The structures of synthetic anilines are listed in Tables 1 and 2.
TEAC assay
ABTS was dissolved in distilled water (38.5 mg in 10 mL) and potassium persulfate (6.90 mg) was added. In order to obtain the radical cation ABTS+', the final solution was left to settle at room temperature in the darkness for 20 hours. An aliquot of ABTS+' was diluted in ethanol to achieve an absorbance of 0.70 ( ± 0.20) at 734 nm. A calibration curve was performed with the reference antioxidant Trolox (dA vs. mmol Trolox), for a decrease of absorbance (dA), produced 6 min after the addition of 30 µL of Trolox solution (0.25-1.98 mM) to 3 mL of the ABTS+' solution. Five solutions of knownconcentrationofeachanilinewere assessed to cause a decrease in absorbance of the ABTS+', such as with the Trolox solutions (dA vs. mmol aniline). The TEAC of anilines was estimated in relation to Trolox, using the relationship between the slopes of the curves of the anilines and that of Trolox (mmol Trolox/ mmol aniline) (15).
EC50 assay
A calibration curve with standard ethanolic DPPH solutions was developed at 2 = 514 nm. The stationary states for five different concentrations of anilines were used to test the antioxidant capacity with 2.5 mL of DPPH solution (90 ± 5 µM) and 0.5 mL of aniline solution. Based on the data obtained (DPPH absorbance vs. time) the graphs from remaining DPPH in stationary state (%) vs. Effective Concentration ((EC), aniline concentration µM/initial DPPH concentration µM) were built. The values of EC50 for anilines were interpolated from these graphs (16).
Dataset preparation and molecular descriptors
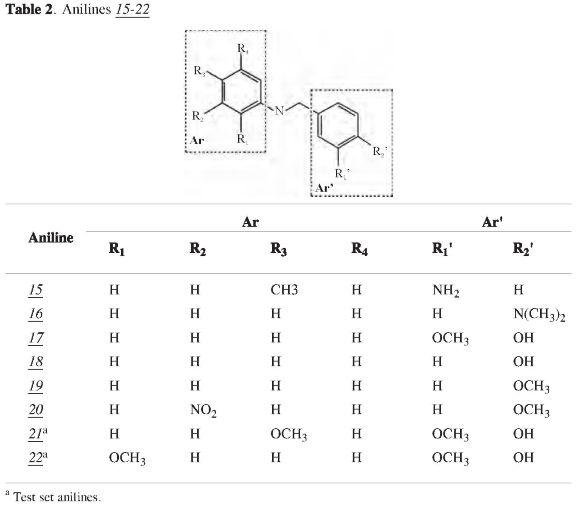
Ab initio, constitutional, geometrical, topological, information, charge distribution descriptors were calculated from anilines 1-12 and 15-20 which were used as the training set, and anilines 13, 14, 21 and 22 which were used as the test set for external validation.
The molecular descriptors HOMO and LUMO energies, Dipolar Moment (DM), EE, IP (-Ehomo), and αzz, were computed with the Gaussian 03W software package using the method and the basis set RHF/CEP-121G (19, 20). This basis set, CEP-121G, was preferred to SDD, LanL2DZ or LanL2MB because the calculated IP of the anilines 23-30 presented greater accuracy (see Table 3). The anilines 23-30 used to choose the basis set with minor relative error were selected among the anilines whose IP in gaseous phase is reported in the Handbook of Chemistry and Physics 1997-1998 (21).
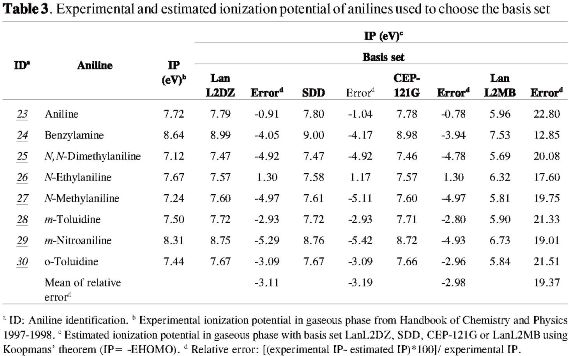
CEP-121G is the combination of a basis set using the compact effective potential (CEP), of the electrons in the core layers, with wave functions of three different sizes for each valence orbital (121, Split Valence) (19, 20). The ab initio effective potentials reduce the difficulties in calculations of species containing heavy atoms (e. g. anilines 7-9 of the training set). The electrons of a molecule are divided into two groups, the core electrons (CE) and valence electrons (VE), so that the energy of the molecule is the sum of the CE and VE energies. In this approach, the CE are treated as a charge distribution that provides an effective repulsive potential V(i) for the movement of the VE. This leads to an effective Hamil
tonian for the VE (H), which is used in the variational integral (19). Additionally, many of the electronic repulsion integrals of core layers are not explicitly represented, since these orbitals change little with the formation of the molecule. The ECP basis sets that can be applied in the Gaussian 03 program include: CEP-121G (Stevens/Basch/Krauss ECP triple-split basis); LanL2MB (STO-3G on the first row and Los Alamos ECP more MBS on Na-Bi); LanL2DZ (D95 on the front row and Los Alamos ECP more DZ on Na-Bi); and SDD (D95V on the first row and Stuttgart/Dresden ECPs on the rest of the periodic table) (19).
Descriptors such as number of atoms (NA), relative number of H atoms (RNH), number of benzene rings (NBR), number of aromatic bonds (NAB), number of single bonds (NSB), molecular weight (MW), Wiener index (WI), information content of order 1 (IC), complementary information content of order 0 (CIC), 3D-Kier & Hall index of order 3 (KH), total charge weighted partial positively charged surface area (PPSA-2), partial negative surface area (PNSA-1), fractional PPSA (FPSA-3), atomic charge weighted partial negative surface area (PNSA-3), were obtained with MOLDES (Tartu State University, Tartu-Stonia), software available for calculating theoretical molecular descriptors (constitutional, geometrical, topological, information and charge distribution).
Chemometric methods
Regression models were developed for the anilines 1-12 and 15-20 (trainingset). Multiple Linear Regression (MLR) analysis between the theoretical descriptors and the antioxidant capacity (TEAC and EC50) was performed with the STATISTICA 6.0 software (Stat Soft, Tulsa, OK, U.S.A.) using the ordinary least squares regression method. Once the models were established, predictions were made for anilines 1-12 and 15-20 (CV-LOO: internal Cross Validation by Leave One Out procedure), and anilines 13,14,21 and 22 (test set for external validation). Model performance was described by parameters related to model predictive capability (Q2CV-LOO, SECV) and fitting power (R2, Error of Estimate (SE), and the Fisher's value (F)). The first mathematical TEAC model was built with 8 descriptors that were selected by backward MLR starting with 14 descriptors. These 14 descriptors had been selected from the 28 descriptors with high correlation with the experimental TEAC values. The latter 28 descriptors were previously selected as the set with mutual correlation coefficients smaller than 0.8, from the total of 87 descriptors computed by the Gaussian 03W and MOLDES packages. A second TEAC model was developed based on 9 principal components (PC), extracted from the forward MLR between 17 PC and the experimental TEAC values. These 17 PCs resulted from principal components analysis (PCA), onthe28 descriptors earlier mentioned. The molecular descriptors used for third and fourth models of TEAC and models of log (1/EC50) were chosenamong abinitio and charge distribution descriptors by random combination.
RESULTS AND DISCUSSION
IP, TEAC and EC50 experimental and predicted values of the anilines 1-22 are shown on the Table 4. The antioxidant capacity evaluated as TEAC and EC50 is related to the electron and proton transferences from the antioxidant species to the ABTS radical cation or DPPH radical, respectively, i. e., the fast oxidation and stability of the antioxidant (15, 16). In the training set (1 -12 and 15-20), only aniline 17(TEAC = 0.86; EC50= 0.23) showed a good reactivity toward radicals because a good antioxidant must have a TEAC value higher than 1 and an EC50 value lower than 1. Therefore, the introduction of a hydroxyl group into the aromatic amine structure enhances its antioxidant performance (reactivity to radicals), probably due to the decrease of IP and BDE (Bond Dissociation Enthalpy), which makes it significantly more reactive in relation to carbon-centered organic radicals. Moreover, replacement of the hydrogen atom of a hydroxyl group (18) with a methyl group (19) drastically decreases the anti-radical capacity (11, 22-26).
The multidimensional models obtained for the prediction of TEAC and EC50 using a training set of 18 anilines and an external evaluation set of 4 anilines (highlighted with the f letter in Table 4), are reported below together with their statistical parameters:
TEAC (1) = (0.49 ± 0.03) + (-2.2 ± 0.5) RNH + (0.9 ± 0.3) WI + (1.5 ± 0.3) CIC + (0.9 ± 0.2) PPSA-2 + (-0.6 ± 0.1) PNSA-3 + (0.27 ± 0.08) DM + (-0.4 ± 0.1)ELUMO + (3.6 ± 0.7)EE

TEAC (2) = (0.49 ± 0.02) + (-0.047 ± 0.005) PC1+ (-0.111 ± 0.005) PC2 + (-0.039 ± 0.008) PC4 + (0.04 ± 0.02) PC5 + (-0.45 ± 0.05) PC8 + (-0.41 ± 0.06) PC9 + (0.33 ± 0.09) PC11 + (-2.3 ± 0.5) PC14 + (-7 ± 2) PC16

TEAC (3) = (0.012 ± 0.003) αzz + (0.009 ± 0.002) PPSA-2 + (0.019 ± 0.004) EE
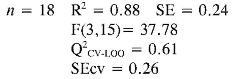
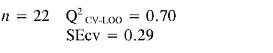
TEAC (4) = (0.18 ± 0.04) αzz + (0.008 ± 0.002) PPSA-2 + (-2.8E-03 ± 6.0E-04) αzz2 + (-9E-05 ± 1.0E-05) EE2 + (1.1E-05 ± 2.0E-06) αzz3
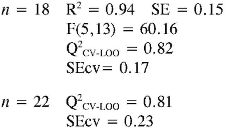
In TEAC model 1, it can be established that anilines 13, 14, 21 and 22 had high deviations between the experimental and predicted values (outliers). This model points out that EE and RNH are important molecular descriptors in predicting TEAC of anilines 1-18. Although, RNH, WI and CIC have a big contribution to the model, this is not enough to think that a physical mechanism is predominant. However, it is important to state that measurement of topological changes in a molecule (related to WI and CIC, among others), can be related to changes in geometry and thus act as a measure of the capacity of the molecular shape to interact with other molecules. Moreover, changes in branching can affect the distribution of electronic charge, which can be reflected as changes in reactivity or polarity (27). In addition, the large difference between R2 and Q2 CV-LOO (n= 18 and n= 22), indicate poor predictive capabilities of the model. Namely, such a model is overfitted, i. e., too many descriptors are introduced into it (28).
In TEAC model 2 with acceptable results in external and internal validations in relation with model 1, it can be seen that anilines 21 and 22 were outliers. The model shows the importance of constitutional, topological, geometric and charge distribution descriptors, related to PC2 and PC7 for predicting the antioxidant capacity by TEAC. Although, principal components as descriptors had a good fit and predictive capabilities by internal validation, their difficult interpretation and poor predictive capabilities by external validation procedure (Q2CV-LOO for n = 22) are the main reasons why the use of this kind of model has few applications in predictability test.
Finally in TEAC models 3 and 4 it is verified that 21 and 22, respectively, were outliers, while none of the predicted data for the set of chemicals evaluated had more than two standard deviations from the experimental value. The differences between R2 and Q2CV-LOO (n = 18 and n = 22), indicated a considerable stability of these models and the possibility of their use in reliable predictions of TEAC. These models validate the importance of quantum descriptors related to αzz, EE and PPSA-2, in predicting the antioxidant capacity. It is important to state that the introduction of nonlinear terms significantly improves the accuracy ofthe TEAC model 4 in relation with model 3 (28), i. e., a polynomial mathematical function is able to describe well the relationship between TEAC and these descriptors.
αzz refers to a molecular quantum descriptor based on the second derivative of the energy in relation to an electric field; it represents the distribution of electrons. In this way, the polarization explained due to resonance, field (dipole) and inductive effects (29), can be one of the main mechanisms of antioxidant capacity as expressed by TEAC. So, soft nucleop-hiles with a low electronegativity are readily polarizable i. e., they are easily oxidized and have high polarizability values (30).
PPSA-2 descriptor is related with polar intermolecular interactions that take place due to charge differences between molecules, and these interactions will be modulated by the amount of each atom exposed to other molecules (31, 32). Then, molecules with a high total charge weighted partial positively charged surface area will have high polar intermolecular interactions and they will be easily oxidized. In addition, a measurement of the distribution of electronic charge can, in some cases, act as a better measure of branching, and therefore of shape, than the typical topological descriptors (27). Finally, the outcomes show that EE is the theoretical descriptor with high contribution to TEAC models. This is a logical correlation; because the energy is a measure of reactivity and molecular stability by the molecular perturbation theory.
The models obtained for the prediction of log (1/EC50) are reported below with their statistical parameters:
Log (I/EC50) (1) = (0.024 ± 0.006) PPSA-2 + (0.044 ± 0.006) EE
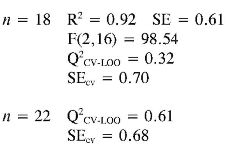
Log (1/EC50) (2) = (-0.013 ± 0.003) αzz + (-1.3E-04 ± 3.0E-05)EE2 + (5.8E-07 ± 7.0E-08) PPSA-23
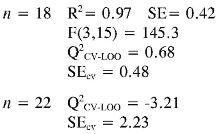
Log (1/EC50) (3) = (-0.017 ± 0.008) αzz + (0.029 ± 0.005) PPSA-2 + (0.031 ± 0.008) EE
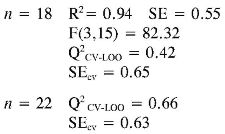
Again the log (1/EC50) models point out the importance of quantum descriptors related to charge distribution (PPSA-2) and electronic energy (EE and αzz) in predicting the antioxidant capacity by EC50. However, the internal and external (Q2CV-Loo for n = 18 and n = 22) validations show that their predictions are a not very reliable, and therefore these models cannot be useful for prediction.
CONCLUSIONS
In this paper, we have analyzed the relationship between the antioxidant capacity evaluated with the TEAC and EC50 assays and theoretical molecular descriptors that are easily and rapidly calculated from the chemical structure of anilines 1-22 with MOLDES and Gaussian 03W packages. Since predictive validation is one way to reliably assess model adequacy for new compounds, in this case only the TEAC model (3) can be quite suitable for prediction purposes. However, it must be underlined that the predicted data must be considered reliable only for those chemicals that fall within the chemical domain on which the model was obtained.
Models for TEAC with good fit and prediction capacity by internal and external validation are obtained using descriptors related with electronic energy (EE and αzz) and charge distribution (PPSA). TEAC models based on constitutional, topological, information content or principal components (PC) as descriptors had in some cases a good fit but showed a poor prediction capacity.
ACKNOWLEDGMENTS
Geovanna Tafurt thanks Colciencias (Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnologia) by her scholarship of Programa de Apoyo a Doctorados Nacionales-2003.
REFERENCES
1. Siraki, A. G.; Chan, T. S.; Galati, G.; Teng, S.; O'Brien, P. N-Oxidation of aromatic amines by intracellular oxidases. Drug metabolism Reviews. 2002. 34 (3): 549-564. [ Links ]
2. Burton, A.; Ingold, K. U.; Walton, J. C. Absolute rate constants for the reactions of primary alkyl radicals with aromatic amines. J. Org. Chem. 1996. 61 (11): 3778-3782. [ Links ]
3. McCowan, J. R.; Yu, M. J.; Phebus, L. A.; Towner, R. D.; Ho, P. P. K.; Keith, P. T.; Luttman, C. A.; Saunders, R. D.; Ruterbories, K. J.; Lindstrom, T. D.; Wikel, J. H.; Morgan, E.; Hahn, R. A. Benzylamine antioxidants: relationship between structure, peroxyl radical scavenging, lipid peroxidation inhibition, and cytoprotection. J. Med. Chem. 1993. 36 (9): 1262-1271. [ Links ]
4. Livingstone, D. Data analysis for chemists. Applications to QSAR and chemical product design. New York: Oxford University Press. 1995. [ Links ]
5. Lien, E.; Ren, S.; Bui, H.; Wang, R. Quantitative structure-activity relationship analysis of phenolic antioxidants. FreeRad. Biol. Med. 1999.26 (5): 285-294. [ Links ]
6. Niculescu-Duvayz, I.; Simon, Z.; Tiriac, S.; Ionescu, A.; Mracec, M.; Voiculetz, N. Quantitative structure-activity relationship (QSAR) applications in chemical carcinogenesis. III. QSAR analysis of some phenolic antioxidants. Neoplasma. 1985. 32 (6): 695-707. [ Links ]
7. Gramatica, P.; Consonni, V.; Pavan, M. Prediction of aromatic amines mutagenicity from theoretical molecular descriptors. SAR QSAR Environ. Res. 2003. 14 (14): 237-250. [ Links ]
8. Ji, H. F.; Zhang, H. Y.; Shen, L. Proton dissociation is important to understanding structure-activity relationships of gallic acid antioxidants. Bioorg. Med. Chem. Lett. 2006. 16 (15): 4095-4098. [ Links ]
9. Reis, M.; Lobato, B.; Lameira, J.; Santos, A. S.; Alves, C. N. A theoretical study of phenolic compounds with antioxidant properties. Eur. J. Med. Chem. 2007. 42 (4): 440-446. [ Links ]
10. Zhang, H. Y.; Wang, L. F. Theoretical elucidation ofstructure-activity relationship for coumarins to scavenge peroxyl radical. J. Mol. Struct. (Theochem). 2004. 673 (1-3): 199-202. [ Links ]
11. Wright, J.; Johnson, E.; di Labio, J. Predicting the activity of phenolic antioxidant: theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001. 123 (6): 1173-1183. [ Links ]
12. Huang, H.; Ou, W.; Zhao, J.; Chen D.; Wang, L. A comparative study of quantitative structure-activity relationship methods based on gallic acid derivatives. SAR QSAR Environ. Res. 2004. 15 (2): 83-99. [ Links ]
13. Heim, K. E.; Tagliaferro, A. R.; Bobilya, D. J. Flavonoid antioxidants: chemistry, metabolism and structure-activity relationships. J. Nut. Biochem. 2002. 13: 572-584. [ Links ]
14. Radomski, J. L. The primary aromatic amines: their biological properties and structure activity relationships. Ann. Rev. Pharmacol. Toxicol. 1979. 19: 129-157. [ Links ]
15. Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Rad. Biol. Med. 1999. 26 (9-10): 1231-1237. [ Links ]
16. Brand-Williams, W.; Cuvelier, M.; Berset, C. Use of a free radical method to evaluate antioxidant activity. Lebensm. Wiss. U. Technol. 1995. 28: 25-30. [ Links ]
17. Antolovich, M.; Prenzler, P. D.; Patsalides, E.; McDonald, S.; Robards, K. Methods for testing antioxidant activity. Analyst. 2002. 127: 183-198. [ Links ]
18. Kouznetsov, V.; Astudillo, L.; Vargas, L. Y.; Cazar, M. E. Synthesis ofsomesecondary aminederivatives bearing a heteroaryl fragment. J. Chil. Chem. Soc. 2004. 49 (4): 319-325. [ Links ]
19. Frisch, M. J.; Trucks, G.W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; et al. Wallingford CT: Gaussian, Inc. 2004. Gaussian 03, Revision C.01. [ Links ]
20. Stevens, W. J.; Basch, H.; Krauss, M. Compact effective potentials and efficient shared-exponent basis sets for the first- and second-row atoms. J. Chem. Phys. 1984. 81 (12): 6026-6033. [ Links ]
21. Lide, D. R.; Frederikse, H. P. R. (Editores). CRC Handbook of Chemistry and Physics. 78th Edition. Boca Raton (USA): CRC Press. 1997. [ Links ]
22. Pratt, D.; di Labio, G.; Brigati, G.; Pedulli, G.; Valgimigli, I. 5-Pyrimidols: novel chain-breaking antioxidants more effective than phenols. J. Am. Chem. Soc. 2001. 123 (19): 4625-4626. [ Links ]
23. Denisov, E. T.; Khudyakov, I. V. .Chem. Rev. 1987. 87 (6): 1313-1357. [ Links ]
24. Ksendzova, G. A.; Sorokin, V. I.; Edimecheva, I. P.; Shadyro, O. I. Reactions of arylamine and aminophenol derivatives, and riboflavin with organic radicals. Free Rad. Res. 2004. 38 (12): 1183-1190. [ Links ]
25. Lucarini, M.; Pedrielli, P.; Pedulli, G. F.; Valgimigli, L.; Gigmes, D.; Tordo, P. Bond dissociation energies of the N-H bond and rate constants for the reaction with alkyl, alkoxyl, and peroxyl radicals of phenothiazi-nes and related compounds. J. Am. Chem. Soc. 1999. 121 (49): 11546-11553. [ Links ]
26. Khlebnikov, A. I.; Schepetkin, I. A.; Domina, N.G.; Kirpotina, L. N.; Quinn, M. T. Improved quantitative structure-activity relationship models to predict antioxidant activity of flavonoids in chemical, enzymatic, and cellular systems. Bioorg. Med. Chem. 2007. 15 (4): 1749-1770. [ Links ]
27. Stanton, D. T. On the physical interpretation of QSAR models. J. Chem. Inf. Comput. Sci. 2003. 43 (5): 1423-1433. [ Links ]
28. Lucic, B.; Trinajstic, N. Multivariate regression outperforms several robustarchitectures ofneuralnetworks in QSAR modeling. J. Chem. Inf. Comput. Sci. 1999.39(1): 121-132. [ Links ]
29. Parr, R. G.; Pearson, R. G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983. 105 (26): 7512-7516. [ Links ]
30. Schultz, T. W.; Carlson, R. E.; Cronin, M. T. D.; Hermens, J. L. M.; Johnson, R.; O'Brien, P. J.; Roberts, D. W.; Siraki, A.; Wallace, K. B.; Veith, G. D. A conceptual framework for predicting the toxicity of reactive chemicals: modeling soft electrophilicity. SAR QSAR Environ. Res. 2006. 17 (4): 413-428. [ Links ]
31. Stanton D. T.; Jurs, P. C. Development and use of charged partial surface area structural descriptors in computer-assisted quantitative structure-property relationship studies. Anal. Chem. 1990. 62 (21): 2323-2329. [ Links ]
32. Stanton, D. T.; Dimitrov, S.; Gran-charov, V.; Mekenyan, O. G. Charged partial surface area (CPSA) descriptors QSAR applications. SAR QSAR Environ. Res. 2002. 13 (2): 341-351. [ Links ]

