










Services on Demand
Journal
Article
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804On-line version ISSN 2357-3791
Rev.Colomb.Quim. vol.39 no.2 Bogotá May/Aug. 2010
MODELAMIENTO POR HOMOLOGÍA DE LA ESTRUCTURA TRIDIMENSIONAL DE LA FOSFOLIPASA A2 CITOSÓLICA PANCREÁTICA DEPENDIENTE DE CALCIO PRESENTE EN Rattus norvegicus
HOMOLOGY MODELLING BY THREE-DIMENSIONAL STRUCTURE OF PHOSPHOLIPASE A2 CYTOSOL-DEPENDENT PANCREATIC CALCIUM IN Rattus norvegicus
MODELAGEM POR HOMOLOGUA DE ESTRUTURA TRIDIMENSIONAL DA ENZIMA FOSFOLIPASE A2 PANCREÁTICAS CITOSSOL CÁLCIO-DEPENDENTE EM Rattus norvegicus
Ricardo Vivas-Reyes1,2, Maicol Ahumedo1, José Cabezas1
1 Programa de Química, Facultad de Ciencias Exactas y Naturales, Universidad de Cartagena, Campus de Zaragocilla. Cartagena, Colombia.
Recibido: 1/12/09 - Aceptado: 10/08/10
RESUMEN
La fosfolipasa A2 dependiente de calcio es de vital importancia en la biosíntesis de mediadores lipídicos, inflamatorios o eicosanoides, tales como: tromboxanos, leucotrienos y prostaglandinas. Para la fosfolipasa A2 dependiente de calcio de 85 kDa presente en Rattus norvegicus, se conoce su estructura primaria (secuencia de aminoácidos), pero a pesar de su relevancia no existen reportes de sus estructuras secundarias y terciarias.
En este estudio se obtuvieron y evaluaron los modelos de estructura terciaria mediante el uso de servidores de modelado y evaluación de proteínas vía Internet para la fosfolipasa A2 dependiente de calcio presente en células beta del páncreas de Rattus norvegicus.
Los modelos 3D obtenidos de la cPLA2 estudiada pueden ser útiles en el diseño racional de experimentos de mutagénesis dirigida, para elucidar y comprender cuál puede ser el mecanismo de funcionamiento de dicha enzima o para el diseño de fármacos que se unan a ella.
Palabras clave: modelamiento, fosfolipasa A2, bioinformática.
ABSTRACT
Calcium-dependent phospholipase A2 (cPLA2) is of vital importance in biosynthesis of lipid, inflammatory or eicosanoid mediators, such as thromboxanes, leukotrienes and prostaglandines. For the 85 KDa cPLA2 present in Rattus norvegicus, its primary structure (AA sequence) is well known, but there is no report of its secundary and tertiary structures.
In this study tertiary structure models of cPLA2 present in pancreatic beta cells of rattus norvegicus were obtained and evaluated using protein modelling and evaluation via internet servers.
The cPLA2 3D models obtained can be useful to the rational design of site-directed mutagenesis experiments, for elucidating and understand which might be the operation mechanism of this enzyme or for the design of drugs which may bind to it.
Key words: modelling, phospholipase A2, bioinformatics.
RESUMO
A Fosfolipase A2 dependente do calcio é de vital importancia na biosíntese de mediadores lipídicos, inflamatorios ou eicosanóides, tais como: tromboxanos, leucotrienos e prostaglandinas. Para a fosfolipase A2 dependente de calcio de 85 kDa presente em rattus norvegicus, se conhece sua estrutura primaria (sequência de aminoácidos), mas nao existem reportes de suas estruturas secundarias y terciarias.
Neste estudo foram obtidos e avaliados os modelos de estrutura terciaria, através do uso de servidores de modelagem e avaliaçao de proteínas, via Internet para a fosfolipase A2 dependente do calcio presente em células betas do páncreas de rattus norvegicus.
Os modelos 3D obtidos da cPLA2 estudada podem ser úteis no desenho racional de experimentos de mutagénese dirigida, para elucidar y compreender qual pode ser o mecanismo de funcionamiento desta enzima ou para o desenho de fármacos que se liguem a ela.
Palavras-chave: modelagem, fosfolipase A2, bioinformática.
INTRODUCCIÓN
El conocimiento de la estructura tridimensional de una proteína es imprescindible si se desea comprender a fondo su mecanismo de funcionamiento o llevar a cabo el diseño racional de fármacos que se unan a ella. Sin embargo, la determinación experimental de estructuras de proteínas es una tarea que a menudo resulta difícil de ejecutar, debido a la imposibilidad de obtener cantidades suficientes de proteína purificada, y también a dificultades durante el proceso de cristalización (grupo espacial y empaquetamiento en la estructura cristalina, presencia o ausencia de ligandos, diferente grado de hidratación, presencia de sales, etc.), o cualquier otro de los muchos problemas técnicos que pueden surgir durante los estudios estructurales (1). Dado el marcado interés que muestran los investigadores por la estructura de las macro-moléculas biológicas, y conscientes de la estrecha relación que existe entre la estructura de una molécula y su función, así como de los grandes avances que se han producido en las técnicas experimentales para el estudio de estructuras, especialmente en las técnicas de cristalización de macromoléculas y complejos de interés biológico, de resolución de estructuras a partir de los patrones de difracción de rayos X y de estudios de macromoléculas en disolución mediante resonancia magnética nuclear (RMN) (1, 2), han aumentado de manera notoria, el número de estudios enfocados al diseño de fármacos basados en la estructura del receptor (métodos directos) y además son el punto de referencia vital en los métodos de modelado por homología de proteínas.
Muchos de los avances en estas técnicas se deben al desarrollo de métodos computacionales específicos de análisis de datos, que han facilitado en gran medida la interpretación de los resultados experimentales. En este estudio nos limitaremos solo al estudio de algunas técnicas bioinformáticas más generales, que han resultado ser de gran utilidad para los investigadores en biología molecular, como son la búsqueda y visualización de estructuras depositadas en las bases de datos, o la modelización molecular, que consiste en predecir de forma teórica la estructura de una macromolécula de la cual se conoce únicamente su secuencia o estructura primaria. La modelación en este caso se ve justificada, ya que de la fosfolipasa A2 dependiente de calcio (cPLA2) presente en Rattus novergicus, se conoce su estructura primaria, pero no existen reportes de sus estructuras secundarias y terciarias. Estos hechos se complementan con las dificultades experimentales existentes para la obtención de estructuras tridimensionales resueltas, y por tanto siempre existe un desequilibrio entre el número de secuencias conocidas y las resueltas experimentalmente. No es sorprendente, por consiguiente, que los métodos de predicción de estructuras de proteínas a partir de su secuencia sean una solución alterna para los estudio de elucidación estructural de macromoléculas, y objeto de un creciente interés (2).
Las fosfolipasas A2 (PLA2) pertenecen a una familia de enzimas que hidrolizan el enlace éster SN-2 de los glicero-fosfolípidos liberando ácidos grasos, principalmente el ácido araquidónico y un lisofosfolípido. Dependiendo de sus isoformas, el sustrato para la PLA2 puede ser fosfatidilcolina, fosfatidiletanolamina, plasmalogenos u otros. El ácido araquidónico sirve como un precursor para la generación de una familia de mediadores lipídicos bioactivos conocidos como eicosanoicos, que incluyen a prostaglandinas, tromboxanos y leucotrienos. Más recientemente, el ácido araquidónico, así como otros ácidos grasos insaturados, ha mostrado realizar funciones importantes como mensajero secundario (1, 3). La fosfolipasa A2 citosólica (cPLA2) hidroliza el enlace SN2-acil éster de los fosfolípidos, y muestra una preferencia por los sustratos que contienen ácido araquidónico.
Se ha reportado que la serina 228 es esencial para la actividad enzimática y funciona como un nucleófilo en el centro catalítico de la enzima (2, 4). La cPLA2 contiene un motivo catalítico ácido aspartico, común para la familia de subtilisina serinas proteasas. La sustitución de alanina por ácido aspartico 549 inactiva completamente la enzima, y sustituciones con ácido glutámico o asparragina reduce la actividad 2.000 y 300 veces, respectivamente. Se encontró también que al sustituir arginina 200 por alanina o histidina, la enzima se inactiva. Dichos resultados están soportados experimentalmente por dicroísmo circular, el cual ha provisto evidencia de que la Asp-549 y la Arg-200 son esenciales para la función enzimática (5).
La clasificación de las fosfolipasas está determinada por el sitio de acción de cada una, su distribución, su modo de acción y su hidrofobicidad. Las fosfolipasas A son acil-hidrolasas y se clasifican de acuerdo con su sitio de acción hidrolítica; por ejemplo, la fosfolipasa A1 hidroliza solo 1-aciléster, mientras que la PLA2 hidroliza solo la 2-aciléster (6). La hidrólisis catalizada por la PLA2 es exergónica y unidireccional, liberando el araquidonato de los fosfolipidos (6). Otras fosfolipasas hidrolizan ambos grupos y se denominan fosfolipasa B; por tanto, se consideran fosfolipasas de gran actividad. Las fosfolipasas C catalizan la rotura del enlace glicerol-fosfato; mientras que la fosfolipasa D puede remover grupos polares básicos. Las fosfolipasas C y D son fosfodiesterasas (6).
MATERIALES Y MÉTODOS
Caracterización de la secuencia
Usando la base de datos UniProtKB se obtuvo la estructura primaria de la cPLA2 presente en los islotes pancreáticos de la Rattus norvegicus (grupo IVA), la cual posee 752 aminoácidos con un peso molecular de 85 kDa, cuyo código de acceso es P50393. Se utilizó el algoritmo de búsqueda BLAST (herramienta de alineamiento básico local) para analizar nuestra secuencia de interés y buscar secuencias de proteínas potencialmente similares a la nuestra, ya que este programa puede usarse gratuitamente desde el servidor del Centro Nacional para la Información Biotecnológica (NCBI). Posteriormente a este alineamiento se analizaron los dominios de la cPLA2 a través del servidor Pfam (7).
El método empleado para la predicción de la estructura secundaria fue: JPRED (10), y se compararon con los resultados de los métodos -SSPRO (9) y HNN (8). Estos métodos clasifican cada aminoácido según tres posibles estados de estructura secundaria: alfa-hélice, hoja-beta u otro. A la predicción de cada residuo se le asigna un índice de fiabilidad, que varía entre 0 y 9, y que se correlaciona con la exactitud de la predicción. Este índice permite identificar las regiones de la proteína que se han predicho con mayor exactitud (11). Se realizó un alineamiento de las secuencias de la cPLA2 utilizando el programa Clustal-W y la base de datos PDB, para encontrar moldes adecuados para la proteína en estudio.
Construcción de los modelos
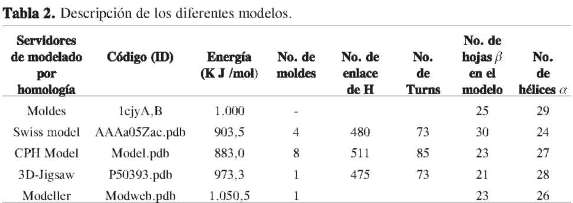
Para motivos de comparación, a partir de la secuencia de la cPLA2 (P50393) se construyeron modelos teóricos usando los servidores Swiss model (12), CPH models (13), 3D-Jigsaw (14), Modweb (15).
Los modelos fueron evaluados en términos estereoquímicas por el programa Whatcheck empleando la interfaz Web del servidor Whatif y por el programa Procheck (16, 17). Además, fueron consideradas las restricciones de los ángulos dihedros psi y phi proporcionados por el diagrama de Ramachadram, a través del servidor imoltalk (18). De igual forma, fue revisada por potenciales energéticos utilizando el programa Prosa 2003, el servidor ProQ que emplea redes neuronales para evaluar la calidad del modelo, basado en contactos intraatómicos y propiedades fisicoquímicas y por último el servidor 3D Evaluation (19, 20).
Caracterización estructural de los modelos
La caracterización de la estructura se realizo a través del programa Promotif (21) que ejecuta una implementación local del algoritmo DSSP (Diccionario estándar de estructuras secundarias) (22). Asimismo, se analiza la superficie y el sitio activo, calculando la accesibilidad de cada unos de los residuos que la componen a partir del programa Mol mol (23).
RESULTADOS Y DISCUSIÓN
Los resultados de los cálculos obtenidos por BLAST muestran que existe una secuencia de proteínas que tienen un alto grado de similitud a la secuencia de aminoácidos de la proteína en estudio, lo que sugiere que esta secuencia pueden ser usada como plantilla, ya que se encontró que posee estructura tridimensional resuelta en el PDB (Protein Data Bank) y su código es 1BCI. Por otro lado, utilizando la opción búsqueda de secuencia disponible en el PDB, se encontraron varias secuencias homólogas a nuestra secuencia problema; un alineamiento secuencial con la proteína blanco fue ejecutado y se encontraron secuencias homólogas con estructura tridimensional resuelta. La secuencia de aminoácidos de cPLA2 presentó un porcentaje de identidad superior al 90% con los mejores moldes encontrados. Con el molde 1CJY se halló una semejanza en la secuencia del 94% sobre 708 AA; con el molde 1RLW se encontró una semejanza del 98% sobre 135 AA, y el molde 1BCI mostró una semejanza en la secuencia del 97% sobre 120 AA. Un alineamiento en pareja (pairwise alignment) se realizó entre nuestra secuencia y el molde 1CJY, ya que esta posee un número de aminoácidos muy parecido al de nuestra secuencia (Figura 1).

El análisis de las similitudes y diferencias en aminoácidos individuales busca inferir relaciones estructurales, funcionales y evolutivas entre las secuencias en estudio. Por tal motivo se puede decir que existen pocas diferencias entre la fosfolipasa 2 humana (lcjy) y la de rata, mostrando una semejanza en la secuencia del 94% y una identidad en la secuencia del 97%. La similitud entre dos secuencies es una variable continua que mide el nivel de coincidencia. La homología se reserva como una variable dicotómica, la cual indica si dos secuencias son homologas o no, dependiendo del grado de similitud y la significancia estadística del alineamiento. Esto deja claro que la fosfolipasa 2 humana (1CJY) es el molde adecuado para realizar el modelado por homología basado en la alta similitud presente entre estas proteínas.
Predicción de la estructura secundaria
La predicción de la estructura secundaria se hizo mediante el uso del servidor JPRED, donde se muestra que la proteína presenta una mayor proporción de loops, lo cual implica una estructura secundaria poco definida (Tabla 1). JPRED posee varios algoritmos, como Jnet que realiza una predicción de la estructura más exacta.


Los dominios se obtuvieron con la ayuda del programa Pfam, donde se encuentra que la cPLA2 blanco P50393 posee dos dominios: uno del tipo C2, el cual se une al calcio, cuya región abarca los aminoácidos que están entre el aminoácido 20 hasta el 106, y un dominio catalítico que comprende los aminoácidos entre el 190 y 675, del cual se encontró que se conserva el motivo lipasa GLSGS (Figura 3).

Se obtuvo el modelo de la cPLA2 usando el servidor de Swiss model empleando los moldes mencionados, y se halló un porcentaje mínimo de identidad de 93,42%, correspondiente al molde 1CJYA, con un máximo de identidad de 97,6%, correspondiente al molde 1BCI; además, los valores de P(N) fueron cercanos a cero, lo cual confirma la semejanza de los modelos con el molde. El modelo obtenido se visualizó usando el programa Deep View, el cual muestra una pequeña proporción de hélices alfa y hojas beta y una alta proporción de estructuras aleatorias.
También se emplearon los programas 3D-Jigsaw, Modeller y CPH model para obtener el modelo 3D. Este último mostró valores de semejanza cercanos al 85%, y además fue visualizado utilizando un límite de confiabilidad de 0,05, mientras los programas 3D-Jigsaw y Modeller mostraron buenos resultados con porcentajes de identidad de 95,7% y 96%, respectivamente (Figura 4). Las principales propiedades de los modelos obtenidos fueron evaluadas y consideran los factores predominantes para la calidad del modelo (Tabla 2).
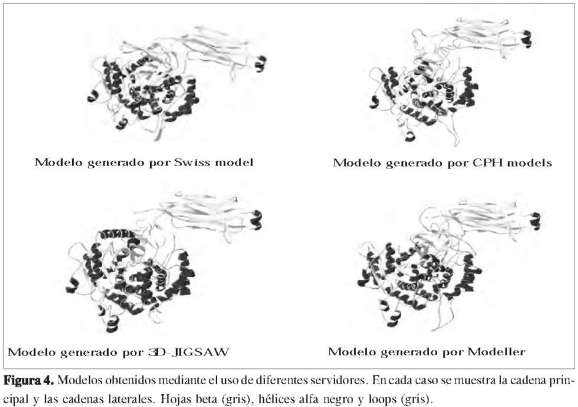
Evaluación de los modelos
Se calculó el RMSD, superponiendo el molde 1CJYA con la secuencia de aminoácidos cPLA2 P50393 modelada por los diferentes servidores de modelado por homología empleando el programa Swiss pdb Viewer (Deep View), haciendo uso de la opción Magic Fit e Interactive Magic Fit del programa, donde se ajustaron los carbonos alfa de los modelos AAAa05Zac.pdb, model.pdb, P50393.pdb y Modweb.pdb con el molde 1CJYA de estructura conocida por cristalografía de rayos X de resolución 2,5 A. De este modo, para modelos que presenten un porcentaje de identidad mayor al 90%, se esperaría que el RMSD calculado de los modelos con respecto al molde 1CJYA sea de 0,5 A. Sobre la base de este hecho se espera que los modelos que presenten un RMSD inferior al valor mencionado con anterioridad sean estructuralmente confiables, como es el caso para los modelos AAAa05Zac.pdb y P50393.pdb (Tabla 3).

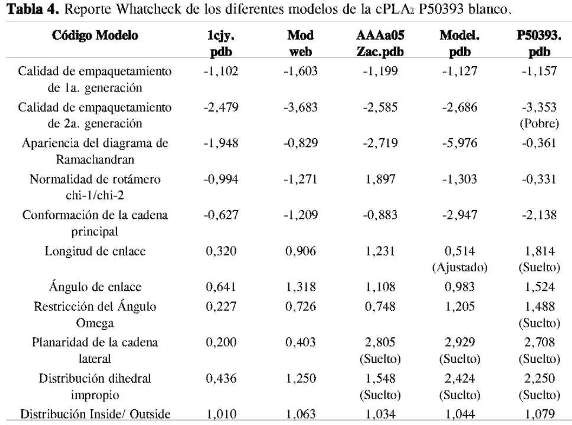
También se utilizó el servidor Whatcheck para evaluar la calidad de los modelos. En esteservidorseconsideraelvalordeZ-value, el cual indica la normalidad de un resultado; este puede ser expresado como un Z-value o Z-score. Este es sólo el número de desviaciones estándar que el resultado se desvía del valor esperado. Una propiedad de Z-values es que la desviación cuadrática media relativa de un grupo de Z-values (el RMS Z-value) esperado es de 1,0. Z-values mayor a 4,0, y menor que -4,0 es muy poco común.
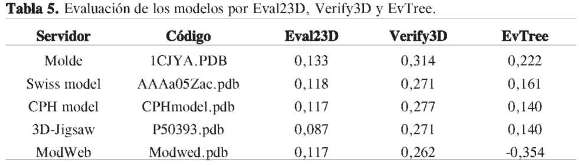
Otro tipo de imprecisión lo constituyen las desviaciones de los valores estereoquímicos ideales para las longitudes y los ángulos de enlaces, de acuerdo con los resultados reportados por Whatcheck para los diferentes modelos (Tabla 4), y el valor esperado para Z-score. El modelo AAAa05-Zac.pdb presenta una mejor precisión con respecto a los otros modelos, aunque para evaluar la calidad de los modelos es necesario ver otros parámetros que sean complementarios, para así determinar si un modelo es útil o simplemente no lo es. Gracias a que existen herramientas computacionales automáticas vía Internet para evaluar la calidad de un modelo, como por ejemplo 3D-Evaluation, el cual hace uso de tres programas de evaluación de estructura basado en potenciales: Eval23D, Verify3D y EvTree, los resultados arrojados por dichos programas muestra cuál de los distintos modelos obtenidos por los diferentes servidores de modelado por homología son los adecuados (Tabla 5).
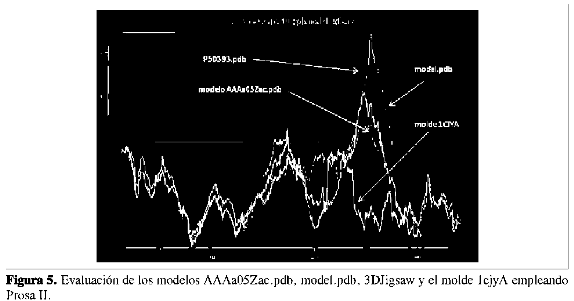
La evaluación de los modelos con Prosa II muestra que la curva del modelo AAAa05Zac.pdb se ajusta más a la curva del molde 1CJYA, con respecto a las otras curvas de los otros modelos model.pdb y P50393.pdb. Al analizar la estructura cristalina de la 1CJY, se encontró que posee regiones sin estructura como lo son las regiones que abarcan los aminoácidos L405-S415, N432-N460 y L498-D539; además, los modelos arrojados por los diferentes servidores de modelado por homología les asignan estructuras a dichas regiones, y este hecho implica que la región que va desde el aminoácido 400 hasta el 575 aproximadamente de los modelos con respecto al molde difiera de manera notable (Figura 5).
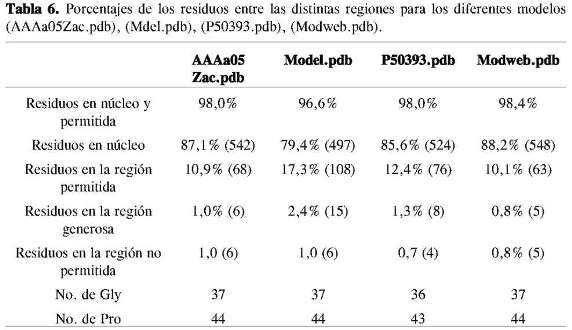
La evaluación de los modelos con Procheck determina la calidad estereoquímica de la estructura de una proteína dada (24). Los valores mayores de 90% para las regiones favorecidas en el diagrama de Ramachandran indican que el parámetro estereoquímico Phi - Psi es ideal para el modelo propuesto (25). Los valores de las regiones favorecidas (núcleo y permitida) para los cuatro modelos (Tabla 6) son mayores a 90%, lo cual pone de manifiesto la calidad de los modelos.
Análisis del mecanismo catalítico
Estudios recientes proponen un mecanismo catalítico de la cPLA2 de 85 kDa. La 1CJYA, que implica el retiro de una capa cerca al sitio activo por la repulsión electrostática no específica, del movimiento del interdominio inducido por el desplazamiento de PtdIns (4,5)P2 y la penetración parcial de la membrana por los residuos hidrofóbicos del dominio catalítico (26), por lo que la cPLA2 blanco modelada se espera que posea un mecanismo similar debido a su alta homología encuanto a secuencia y estructura molecular.
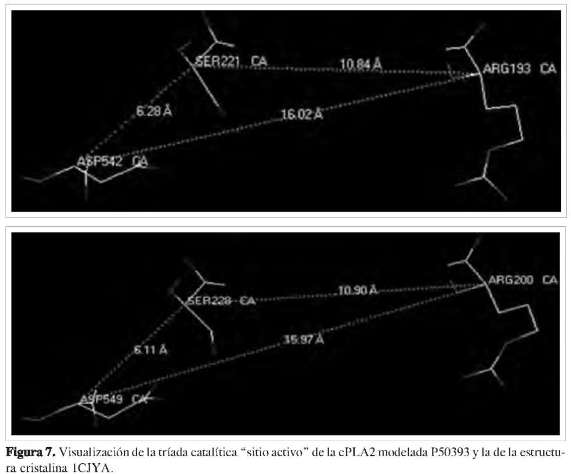
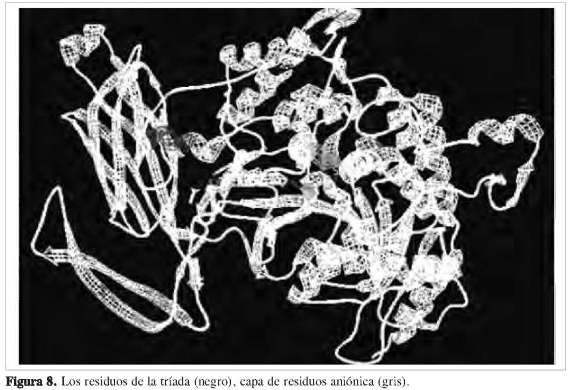
Estudios previos demostraron que la enzima de 85 kDa cPLA2 posee una tríada catalítica situada en una hendidura profunda en el centro de un embudo predominante hidrofóbico que rompe selectivamente el araquidonil de los fosfolípidos, la serina 228, el ácido aspártico 549 y la arginina 200 (27) (Figura 7). Se determinó que dichos residuos de aminoácidos se encuentran en el interior o núcleo de la estructura de la proteína (Figura 6). Al hacer un análisis de superficie ASA (accesibilidad del área superficial) empleando el programa Mol- Mol, los resultados muestran que los residuos de aminoácidos que presentan una accesibilidad igual o mayor a 30% se encuentran en la superficie de la estructura, y los restantes en el núcleo de la proteína (28). La estructura revela una tapa flexible (Figura 8) que debe moverse para permitir el acceso del sustrato al sitio activo, explicando así la activación interfacial de esta importante lipasa. La cPLA2 de 85 kDa media el lanzamiento agonista-inducido del ácido araquidónico en muchos modelos de la célula, incluyendo macrófagos peritoneales del ratón. La cPLA2 es regulada por un aumento de calcio intracelular, el cual se enlaza a un amino-terminal del dominio C2 e induce su desplazamiento sobre la membrana nuclear y el retículo endoplasmático. La fosforilación de la cPLA2 en S505 por la proteína kinasa mitogen-activados (MAPK) también contribuye a la activación. Para que la cPLA2 pueda cumplir su función catalítica, debe cambiar su conformación estructural de tal modo que los residuos que hacen parte del centro catalítico queden expuestos, para así estar lo más próximo al sustrato y poder interaccionar con el fosfolípido de la membrana y liberar el ácido araquidónico.
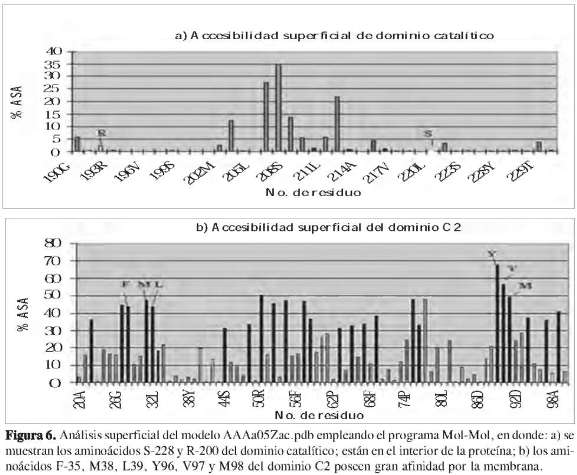
Se encontró teóricamente que los residuos que conforman la tríada catalítica (R-193, S-221 y D-542) se encuentran en el interior (núcleo) de la estructura de la proteína. Además se observa que esta región está ubicada en el interior de la proteína modelada (Figura 6a).
Reportes anteriores muestran que los residuos hidrofóbicos (F-35, M-38 y L-39) están expuestos en la cara de un segmento helicoidal y los residuos (Y-96, V-97 y M98) están expuestos en los ápices de un loop. Estos residuos del dominio C2 pueden contribuir significativamente a enlazarse con la membrana.
Dichos residuos están en la superficie de la proteína (Figura 6b).
La información obtenida a partir de los resultados de BLAST permite explicar que los modelos construidos serán estadísticamente confiables, dado que se considera que las técnicas de modelado por homología son aplicables cuando la proteína problema es similar a una de estructura tridimensional conocida, y su identidad a nivel de secuencia entre ambas proteínas sea mayor de 30%. Esto último permite afirmar que las plantillas utilizadas son idóneas para construir un modelo 3D. Además de estadísticamente confiable, es un modelo estructuralmente adecuado para servir de receptor al realizar estudios de acoplamiento molecular.
La predicción de estructura secundaria hecha para cPLA2 utilizando los métodos HNN, SSPRO y JPRED indican que cPLA2 es una proteína con una estructura secundaria formada principalmente por una conformación de alfa-hélices, con una pequeña proporción de hoja-beta y una cantidad muy considerable de estructura no regular. Esta estructura se obtuvo haciendo un consenso entre los tres tipos de métodos utilizados para hacer dicha predicción (Figura 2).
El análisis de dominios de cPLA2 mostró que en el dominio C2, el cual se une al calcio, cuya región abarca los aminoácidos que están entre el aminoácido 20 y el 106, la cPLA2 es activada por niveles bajos de calcio, pero el calcio no está involucrado en la catálisis, sino que es requerido para enlazarse con la membrana, en donde los aminoácidos de unión con el calcio son: D-40, T41, D43, N65, D93, A94, N95 (29). En el otro dominio, un dominio catalítico que comprende los aminoácidos entre el 190 y el 675, del cual se encontró que se conserva el motivo lipasa GLSGS, revela que la serina 228 es un centro catalítico, y el mecanismo catalítico de la cPLA2 es similar a otras lipasas que contienen esta tríada catalítica (30).
En general, la evaluación hecha por los diferentes servidores de evaluación de modelos como Whacheck, Procheck y 3D-Evaluation, permiten inferir que dichos modelos son aptos y útiles para comprender a fondo su función catalítica. Si se observa la aplicabilidad de los modelos obtenidos y los altos grados de identidad (mayor del 90%), de las secuencias de los moldes y la proteína en blanco, se pueden encontrar inhibidores artificiales que interaccionen con la proteína modelada.
Se observa, igualmente, que existen diferencias entre los resultados obtenidos por cada servidor de evaluación de los modelos construidos por los diferentes servidores de modelado por homología. Estas diferencias conducen a escoger con qué servidor de modelado trabajar para obtener buenos modelos de proteínas. Por último, la calidad de los modelos comparativos de proteínas sólo puede evaluarse a posteriori, porque, a diferencia de las técnicas experimentales, normalmente no intervienen en su construcción datos obtenidos en el laboratorio que sirvan de control.
CONCLUSIONES
Los servidores de modelado por homología, como: Swiss model, 3D Jigsaw, CPH model y Modeller, entre otros, resultan ser herramientas computaciona-les idóneas para el diseño estructural de proteínas, siempre que en las bases de datos de proteínas exista una plantilla o molde con un porcentaje de identidad muy alto y que abarque toda la longitud de la proteína.
Swiss model aventaja a los otros servidores gracias a que puede construir modelos de forma automática. Además, es de muy fácil manejo: solo se necesita que el investigador envíe la secuencia, y en pocos minutos le devuelve un modelo de la proteína. Pese a esta automatización, aunque puede ser útil para generar ideas, se recomienda hacer ciertos ajustes al modelo antes de proseguir a trabajar con él.
Los servidores de evaluación de proteínas, como: Procheck, Whatcheck, 3D-Evaluation, Prosa II, resultan idóneos para evaluar la calidad de los modelos obtenidos para la cPLA2 estudiada.
Los modelos 3D obtenidos de la cPLA2 estudiada pueden ser útiles en el diseño racional de experimentos de muta-génesis dirigida, para elucidar y comprender cuál puede ser el mecanismo de funcionamiento de dicha enzima o para el diseño de fármacos que se unan a ella, así como también para hacer estudios de interacción ligando-proteína, proteína-proteína, y por último realizar la caracterización estructural del modelo.
REFERENCIAS BIBLIOGRÁFICAS
1. Branden, C.; Tooze, J. Introduction to Protein Structure. New York: Garland. 1991. [ Links ]
2. Tithof, P. K.; Peters-Golden, M.; Ganey, P. E. Distinct phospholipases A2 regulate the release of arachidonic acid for eicosanoid production and superoxide anion generation in neutrophils. J Immunol. 1998. 160 (2): 953-960. [ Links ]
3. Sharp, J. D.; White, D. L.; Chiou, X. G.; Goodson, T.; Gamboa, G. C.; McClure, D. Intracellular Ca2 + , inositol 1,4,5-trisphosphate and additional signalling in the stimulation by platelet-activating factor of prostaglandin E2 formation in P388D1 macrophage-like cells. BiochemJ. 1994. 298 (3): 543-551. [ Links ]
4. Richard, T.; Pickard, X.; Grace, C.; Beth, A. S.; Michael, R. D.; Paul, A. H.; Luoise, T.; Ying, K. Y.; Laure, J. R.; Edward, A. D.; Ruth, M. K.; John, D. S. Identification of essential residues for the catalytic function of 85-kDa Cytosolic phospholipase. J. Bio. Chem. 1996. 271: 19225-19231. [ Links ]
5. Hicks-Gómez, J. Bioquímica. Gac MedMex. 2001. 137(5): 112-117. [ Links ]
6. Roskoski, R. Jr.; Ritchie, P. Time-Dependent inhibition of protein farnesyltransferase by a benzodiazepine peptide mimetic. Biochemistry. 2001.40(31): 9329-9335. [ Links ]
7. Robert, F.; Jaina, M.; Benjamín, B.; Sam, J.; Volker, H., et ál. Pfam: clans, web tools and services. Nucleic Acids Research, 2006 Database Issue 34: 247-251. http://www.sanger.ac.uk/Software/Pfam/ [ Links ]
8. Guermeur, Y. (1997). Combinaison de classifieurs statistiques, Application a la prediction de structure secondaire des proteins. PhD Thesis, Université Paris 6. [ Links ]
9. Baldi, P.; Brunak, S.; Frasconi, P.; Soda, G.; Pollastri, G. Exploiting the past and the future in protein secondary structure prediction. Bioinformatics. 1999. 15: 937-946. [ Links ]
10. Cuff, J. A.; Clamp, M. E.; Siddiqui, A. S.; Finlay, M.; Barton, G. J. JPred: a consensus secondary structure prediction server. Bioinformatics. 1998. 14: 892-893. [ Links ]
11. Rost, B. Review: protein secondary structure prediction continues to rise. J Struct Biol. 2001. 134: 204-218. [ Links ]
12. Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M. C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Research. 2003. 31: 3381-1185. http://swissmold.expasy.org//SWISS-MODEL.html [ Links ]
13. CPHmodels 2.0: X3M a Computer Program to Extract 3D Models. O. Lund, M. Nielsen, C. Lundegaard, P. Worning. http://www.cbs.dtu.dk/services/CPHmodels/ [ Links ]
14. Bates, P. A.; Kelley, L. A.; MacCallum, R. M.; Sternberg, M. J. E. a. Enhancement of Protein Modelling by Human Intervention in Applying the Automatic Programs 3D-JIGSAW and 3D-PSSM. b. Proteins: Structure, Function and Genetics, 2001. Suppl. 5: 39-46. [ Links ]
15. Bates, P. A.; Sternberg, M. J. E. (1999). Model building by comparison at CASP3: Using expert knowledge and computer automation. c. Proteins: Structure, Function and Genetics, Suppl 3:47-54 d. Contre-ras-Moreira,B., Bates,P.A. Domain Fishing: a first step in protein comparative modelling. Bioinformatics. 2002. 18: 1141-1142. http://www.bmm.icnet.uk/~3djigsaw/html/form_v2.0_maestro.html [ Links ] [ Links ]
16. Pieper, U.; Eswar, N.; Davis, P.; Braberg, H.; Madhusudhan et al. MODBASE: a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res. 34: 2006 Database issue: 291-295. http://alto.compbio.ucsf.edu/modwebcgi/main.cgi [ Links ]
17. Laskowski, R. A.; McArthur, M. W.; Moss, D. S.; Thornton, J. M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993. 26: 283-291. http://Bitech.embl-ebi.ac.uk:8400/ [ Links ]
18. Diemand, V.; Scheib, H. iMolTalk: an interactive, internet-based protein structure analysis server. Nucleic Acids Research. 2004. (1) 32: W512-W516. Web Server issue. http://i.moltalk.org/iMolTalk.cgi [ Links ]
19. Sippl, M. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993. 4: 355-362. [ Links ]
20. Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Science, 2003. 12: 10731086. http://bioserv.cbs.cnrs.fr/HTML_BIO/frame_valid.html [ Links ]
21. Hutchinson, E.; Thornton, J. PROMOTIF-A program to identify and analyze structural motifs in proteins. Prot. Sci. 1996. 5: 212-220. [ Links ]
22. Kabsch, W.; Sander, C. Dictionary of protein secondary structures: pattern recognition ofhydrogen-bonded and geometrical features. Biopolymers. 1983. 22: 2577-2637. [ Links ]
23. Koradi, R.; Billeter, M.; Wuthrich, K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996.14: 51-55. [ Links ]
24. Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Science. 2003. 12: 1073-1086. http://bioserv.cbs.cnrs.fr/HTML_BIO/frame_valid.html [ Links ]
25. Guex, N.; Peitsch, M. C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modelling. Electrophoresis. 1997. 18: 2714-2723. [ Links ]
26. Morris, A. L.; MacArthur, M. W.; Hutchinson E. G.; Thornton, J. M. Stereochemical quality of protein structure coordinates. Proteins. 1992. 12: 345-364. [ Links ]
27. Le Grand, M.; Merz, Jr. K. M. Optimization. "Application of the Genetic Algorithm to the Minimization of Potential Energy Functions". S. J. 1993. 3: 49-66. [ Links ]
28. Dessen, A.; Tang, J.; Schmidt, H.; Stahl, M.; Clark, D.; Seehra, J. et al. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell. 1999. 97: 349-360. [ Links ]
29. Richard, T.; Pickard, X.; Grace, C.; Beth, A. S.; Michael, R. D.; Paul, A. H. et al. Identification ofessential residues for the catalytic function of 85-kDa Cytosolic phospholipase. J. Bio. Chem. 1996 . 271: 1922519231. [ Links ]
30. Laskowski, R. A.; Luscombe, N. M.; Swindells, M. B.; Thornton, J. M. Protein clefts in molecular recognition and function. Prot. Sci. 1996. 5: 2438-2452. [ Links ]

