










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804On-line version ISSN 2357-3791
Rev.Colomb.Quim. vol.40 no.2 Bogotá May/Aug. 2011
OPTIMIZACIÓN DEL MÉTODO DE MICROEXTRACCIÓN EN FASE SÓLIDA ACOPLADA A CROMATOGRAFÍA GASEOSA PARA ANÁLISIS DE METANOL EN AGUA
OPTIMIZATION OF SOLID PHASE MICROEXTRACTION METHOD COUPLED TO GAS CHROMATOGRAPHY FOR ANALYSIS OF METHANOL IN WATER
OTIMIZAÇÃO DO MÉTODO DE MICROEXTRAÇÃO EM FASE SÓLIDA ACOPLADA Á CROMATOGRAFIA GASOSA PARA ANÁLISE DE METANOL EM ÁGUA
Fidel Granda1,2, Sergio Valencia1, Juan M. Marín1, Gloria Restrepo1.
1 Grupo Procesos Físico-Químicos Aplicados, Universidad de Antioquia, calle 67 53–108, bloque 18, oficina 411, A.A. 1226, Medellín (Colombia).
Recibido: 21/11/10 - Aceptado 05/08/11
RESUMEN
Se optimizó la técnica de microextracción en fase sólida acoplada a cromatografía gaseosa con detector de ionización de flama (HS-SPME/GC-FID por su nombre en inglés) para análisis de metanol en agua utilizando diferentes microfibras, una fibra de poliacrilato y una fibra de polidimetilsiloxano y una columna RTX-5. Las variables evaluadas fueron tiempo de absorción del contaminante en la fibra, volumen libre en el vial, tiempo y temperatura de desorción y efecto de la agitación y presencia de sal en el sistema. De acuerdo con los resultados, el tiempo óptimo de absorción es 15 minutos, el volumen de muestra es 5 mL. Se encontró que existe una íntima relación entre la temperatura de desorción y el área del pico cromatográfico, y una correlación R2 = 0,99965 para la curva de calibración elaborada en un rango entre 0 y 50%, considerando el origen, con un límite de detección de 0,01% y un límite de cuantificación del 0,04%.
Palabras clave: Análisis, medición, metanol, aguas residuales, SPME, poliacrilato, GC - FID.
ABSTRACT
The technique head space-solid phase micro extraction with gaseous chromatography by using flame ionization detector (HS-SPME/GC-FID) for analysis of methanol in water was optimized using different microfibers, a polyacrylate fiber and a polydimethylsiloxane fiber and a RTX-5 column. The evaluated variables were time of absorption of the pollutant on the fiber, free volume in the vial (free space), time and temperature of desorption as well as effect of agitation and presence of salt in the system. According to the results, the optimal absorption time is 15 minutes, the optimal volume of sample is 5 mL. A close relationship between the temperature of desorption and the chromatographic peak area was found, and a correlation R2 = 0.99965 was obtained for the calibration curve in a range between 0 and 50%, considering the origin, with a detection limit of 0.01% and a quantification limit of 0.04%.
Key words: Analysis, measurement, methanol, waste water, SPME, polyacrylate, GC-FID.
RESUMO
A técnica microextração em fase sólida acoplada a cromatografia gasosa com detector de ionização de chama (HS-SPME/GC-FID por seu nome em Inglês) foi otimizada para análise de metanol em água através de diferente micro fibra, uma fibra de poliacrilato e uma fibra de polidimetilsiloxano e uma coluna de RTX-5. As variáveis avaliadas foram tempo para absorver o poluente no volume de fibra livre no frasco, tempo e temperatura de dessorção e efeito da agitação e da presença de sal no sistema. De acordo com os resultados, o tempo de absorção ideal é 15 minutos, o volume da amostra é 5 mL. Descobrimos uma íntima relação entre a temperatura de dessorção e área do pico cromatográfico, e uma correlação R2 = 0,99965 para a curva de calibração desenvolvida em uma escala entre 0 e 50%, considerando a origem, com um limite de detecção de 0,01% e um limite de quantificação de 0,04%.
Palavras-chave: Análise, medição, metanol, água residuária, SPME, poliacrilato, CG - FID.
INTRODUCCIÓN
Para la detección de metanol en diversos medios (agua, aire o suelos) se han desarrollado varios métodos de análisis con diferentes procedimientos de concentración y extracción de la fase acuosa, como la extracción en fase líquida (1, 2), para analizarlo principalmente por cromatografía. A diferencia de los métodos de extracción mencionados, la microextracción en fase sólida (SPME por sus siglas en inglés), reportada por Pawliszyn y colaboradores en 2004 (3), no utiliza ningún solvente para la extracción. Esta técnica consta de dos pasos: extracción y desorción del analito. En el proceso de extracción, se coloca una fibra en una fase líquida o gaseosa del sistema. El analito es retenido en la fibra dependiendo de los coeficientes de distribución; seguidamente, la fibra es transferida a un cromatógrafo para la desorción, separación y cuantificación del analito (4,5).
La SPME es por naturaleza una técnica de equilibrio basada en la partición del soluto entre la fibra y la matriz (acuosa o gaseosa); de esta forma, el analito no se extrae del todo de la matriz. Por tanto, las muestras son analizadas después de alcanzar el equilibrio o en un tiempo específico antes de alcanzar el equilibrio con un cuidadoso control del tiempo y de la temperatura (6).
La SPME es una técnica con alta sensibilidad, con límites de detección en µg L-1, buena linealidad y repetitividad. La extracción se puede hacer directa o indirectamente, esto es, con la fibra en contacto con la solución o en el volumen libre del vial que contiene la muestra (head space, HS). Para analitos con alta volatilidad, es preferible el muestreo en HS debido a que los tiempos de equilibrio son más cortos, comparados con la extracción directa; además de proteger la fibra (3). En la SPME, el tiempo de extracción y el número relativo de moléculas extraídas en distintos tiempos es independiente de la concentración del analito, mientras que el número absoluto de moléculas extraídas en diferentes tiempos es linealmente proporcional a la concentración del analito (7). Para mejorar la velocidad, la sensibilidad y la selectividad de la SPME, se estudian parámetros como temperatura de la muestra, agitación, fibra utilizada, adición de sal, efecto del pH y extracción directa o indirecta (7, 8).
En este trabajo se optimizó la medi-ción de metanol en aguas preparadas en el laboratorio vía microextracción en fase sólida en espacio vacío (HS-SPME) usando dos fibras poliméricas, seguida de cromatografía gaseosa con un detector de ionización de llama (GC-FID).
MATERIALES Y MÉTODOS
El análisis del metanol se realizó mediante cromatografía gaseosa con un detector de ionización de llama (GC-FID), usando un cromatógrafo Agilent Tecnologies 7890 con las siguientes condiciones: temperatura en el inyector a 260 °C con relación de Split 60:1, flujo en el inyector de H2 a 2 mL min-1, temperatura del detector 350 °C, columna RTX-5, 5% difenil-95% poli siloxano (60 m x 0,25 mm d.i. x df 0,25 µm). El programa de temperatura fue: 80 °C durante 1 min, 10 °C min-1 hasta 120 °C, flujo por la columna 2 mL min-1 con una velocidad lineal de 33.169 cm min-1. Para la HS-SPME se utilizaron dos fibras: una de 85 µm de poliacrilato, la cual denominaremos fibra blanca (Supelco), y una de 75 µm de polidimetilsloxano, la cual denominaremos fibra negra (Supelco), ambas acondicionadas por una hora a 280 °C. Para las determinaciones de metanol (grado analítico Merck) se prepararon soluciones en aguas ultra pura (mili Q Millipore) en un rango de composiciones entre 0 y 50%. La muestra con el metanol se inyectó usando viales de 15 mL (Supelco). Además, se determinó el efecto de la agitación y de la adición de NaCl (99,5% Merck) en la absorción del metanol en la fibra.
Para el proceso con HS-SPME/GC-FID las soluciones se transfirieron a viales de 15 mL usando micropipetas; la fibra se colocó en el espacio vacío del vial sin contacto con la solución. Con el fin de establecer las condiciones óptimas para el procedimiento de HS-SPME/GC-FID se evalúo la influencia de los siguientes parámetros: volumen de muestra, tiempo de extracción, tiempo y temperatura de desorción, agitación de la muestra y presencia de sal (en nuestro caso NaCl) en la variación de los valores de área de pico utilizando una solución de 50% v/v. También se evaluó la sensibilidad del sistema estableciendo la relación señal-ruido instrumental.
RESULTADOS Y DISCUSIÓN
Determinación del tiempo de extracción
El tiempo de extracción es uno de los factores más importantes en la mayoría de los procesos de extracción, ya que influye en la precisión de los resultados. El tiempo de equilibrio es definido como el tiempo después del cual la cantidad de analito extraído permanece constante (9) y depende del tipo de propiedades del analito, de la muestra y de la fibra (10). En la Figura 1 se observa que la fibra negra tiene una mejor respuesta para las concentraciones evaluadas que la fibra blanca, debido a las características de las fibras utilizadas.
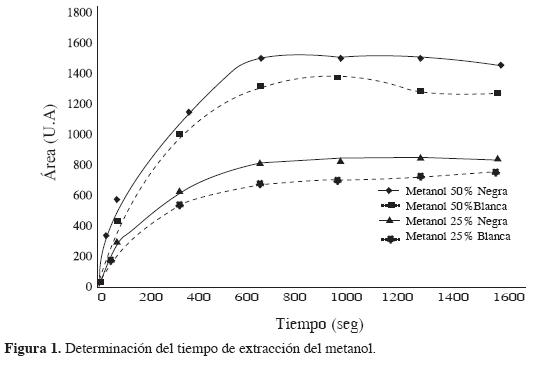
Además, se aprecia la dependencia de las áreas de los picos del metanol con el tiempo en el rango 0-1500 s (0-25 min) con un volumen de 1 mL de muestra (volumen libre 14 mL), temperatura de desorción de 260 °C para soluciones de metanol del 25 y 50% para ambas fibras. Los equilibrios de absorción en ambas fibras se completan en tiempos similares en las concentraciones evaluadas (alrededor de 900 segundos). Este tiempo es independiente de la concentración, mientras que el factor realmente influenciado por la concentración del analito es el área cromatográfica, por las saturaciones de la fibra con el analito, resultado de los dos equilibrios presentados en este tipo de sistemas (líquido-gas y gas-sólido).
Efecto del volumen de muestra y del volumen libre en el vial
Para una alta sensibilidad en la extracción en HS-SPME, el volumen de la fase gaseosa debe ser minimizado para concentrar el analito antes de que se difunda hacia la fibra (11,12). Para determinar la variación de las áreas de los picos con el volumen se usaron viales de 15 mL y volúmenes de muestra incrementadas desde 1 mL hasta 6 mL (volumen libre entre 14 y 9 mL), tiempo de extracción de 15 min y temperatura de desorción de 260 °C. En la Figura 2, al igual que en el caso anterior, la mejor respuesta corresponde a la fibra negra.
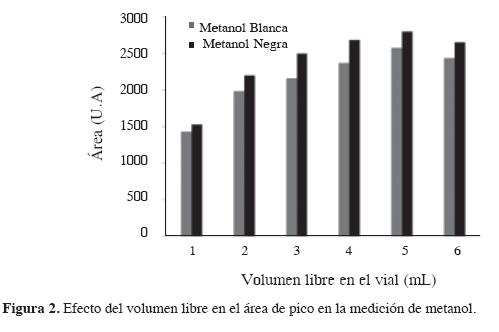
Asimismo, se observa que el equilibrio y la mejor lectura de la concentración del analito en la muestra para ambas fibras se alcanzan con un volumen de muestra de 5 mL (10 mL volumen libre).
Efecto del tiempo de desorción
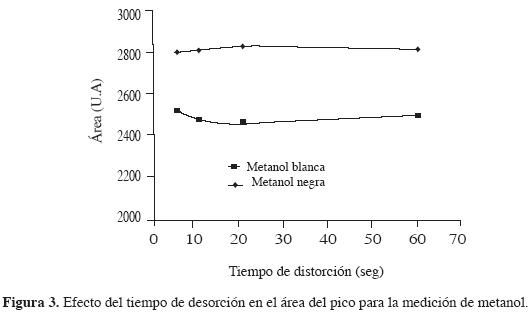
Para determinar la influencia del tiempo en la desorción, los experimentos se realizaron incrementando el tiempo desde 5 s hasta 60 s, utilizando volúmenes de 5 mL de muestra (volumen libre 10 mL), tiempo de extracción de 15 minutos y una temperatura de desorción de 260 °C. Como en los casos anteriores, en la Figura 3 se observa mejor desempeño de la fibra negra. Además, se puede observar que el tiempo de desorción no es un factor relevante en la determinación de la concentración del analito, ya que, por ser una molécula de alta volatilidad, se desorbe rápidamente debido a la temperatura en el detector.
Efecto de la temperatura de desorción
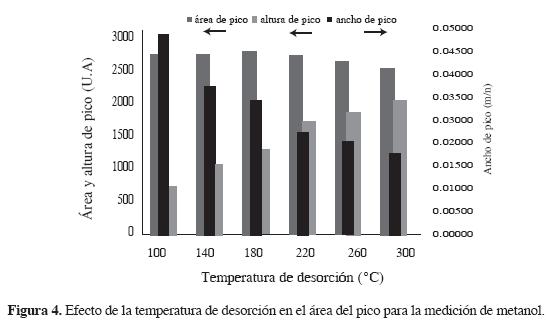
La temperatura de desorción del analito en la fibra en el puerto de inyección del sistema cromatográfico tiene una alta influencia en la forma del pico cromatográfico, como se observa en la Figura 4 (la cual fue realizada para la fibra blanca), donde el área cromatográfica no cambia significativamente, pero sí su conformación. Para temperaturas de desorción bajas se obtienen picos de poca altura, pero muy anchos. A medida que aumenta la temperatura, el ancho del pico disminuye y aumenta su altura, obteniéndose picos más definidos que mejoran la linealidad de analito a bajas concentraciones. Con la forma de los picos obtenidos a temperaturas cercanas a la máxima posible en el puerto de inyección del sistema cromatográfico se muestra que el analito se desorbe más rápidamente a estas temperaturas. Por tanto, se recomienda trabajar a temperaturas cercanas a la máxima a la que se puede someter este tipo de fibra.
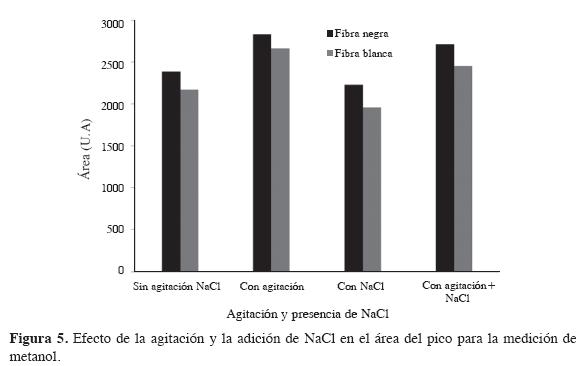
Efecto de la presencia de NaCl y la agitación
Mejorar la fuerza iónica mediante la adición de una sal en las muestras puede incrementar la recuperación de la extracción; esto se conoce como efecto salting out. La agitación mecánica es necesaria para incrementar la difusión de los contaminantes, lo cual es insignificante en muestras acuosas; sin embargo, la capacidad de difusión disminuye significativamente por la presencia de una delgada capa de agua alrededor del recubrimiento de la fibra, conocida como capa límite Prandtl. Cuando esta capa es suficientemente gruesa, se puede reducir la difusión de los contaminantes a través de ella y, por tanto, se prolongan los tiempos de equilibrio en la toma de la muestra (13).
Se realizó un análisis del efecto de la agitación y la presencia de NaCl en el sistema en los valores del área de pico cromatográfico para ambas fibras. En la Figura 5 se observa que, para una agitación de 250 rpm (agitador magnético), el área del pico cromatográfico aumenta, mientras que al agregar NaCl (25% m/v) al sistema (12,13), disminuye el área de pico. Dado lo anterior, en el caso del análisis de metanol vía HS-SPME/GC-FID, el aumento del área cromatográfica al ser agitado el sistema no es tan significativo para seguir utilizándolo, ya que eso incrementaría el costo de los análisis. También se aprecia la influencia negativa de la presencia de NaCl en el sistema; por consiguiente, se concluye que no es necesario agregar sales en los análisis de metanol por este método. El efecto combinado de la agitación y la presencia de sal apantallan el efecto positivo de la agitación con el efecto negativo de la presencia de sal.
Validación de la técnica HS-SPME/GC-FID
Con las condiciones óptimas para la HS-PME -tiempo de extracción 15 min, volumen de muestra 5 mL (espacio vacío 10 mL) y temperatura de desorción 260 °C-, se preparó una curva de calibración graficando las áreas del analito contra el porcentaje de metanol correspondiente a cada área cromatográfica (Aanalito vs %analito) en un rango entre 0 y 50% (vlv) de metanol (los análisis se hicieron por duplicado). La linealidad de la curva se obtuvo por medio de una regresión lineal considerando el origen (punto 0,0). Para comprobar si existe correlación estadísticamente significativa entre la concentración y el área de pico, se aplicó la prueba de hipótesis para la pendiente, con H0: b = 0 y HA: b ≠ 0. También se evaluó la linealidad (r2), la precisión en términos del porcentaje de desviación estándar relativa (% RSD), la exactitud, la recuperación (REC) y los límites de detención (LOD) y cuantificación (LOQ) basados en la relación señal-ruido (S/N) de 3 y 10, respectivamente. También, se evaluaron la precisión y la exactitud del método.
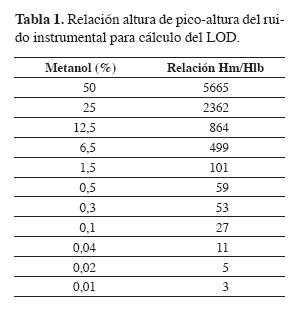
En la Tabla 1 se observan los valores de las relaciones entre las alturas de los picos cromatográfico (Hm) y la altura del ruido instrumental (Hlb) para uno de los porcentajes de metanol medidos. Se halló que el LOD es 0,01% de metanol en una muestra dada. En la Tabla 2 se observa que, para la curva de calibración, el tobservado ⁄ > t(a/2,n-2); por tanto, se rechaza la hipótesis nula acepta la hipótesis alternativa: existe una relación significativa entre la concentración y el área de pico.
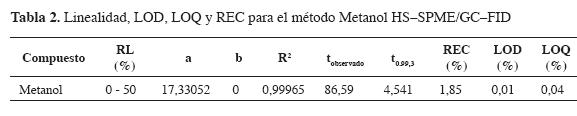
La repetibilidad del método HS-SPME/GC-FID se expresó en el porcentaje de desviación estándar relativa (% RSD). Se utilizó un estándar de 50% de metanol; los análisis de realizaron cinco veces. En la Tabla 3, encontramos los valores de la precisión y la exactitud del método, y vemos que son satisfactorios. La precisión está reflejada en el bajo coeficiente de variación, que para la SPME son permitidos hasta del 12%; para la exactitud, el valor obtenido de tobservado es menor que la t de Student (14) en el intervalo de confianza del 99% y n - 2 grados de libertad. Esto determina que el método tiene la exactitud requerida para el intervalo de confianza en el rango de porcentajes utilizado.

De acuerdo con los resultados de las Tabla 1 y Tabla 2, los límites de detección (0,01%) y cuantificación (0,04%) encontrados para el método optimizado permiten un gran número de mediciones de metanol, debido a que el rango empleado fue bastante grande (en el orden de porcentajes); además, se observa una correlación lineal entre las áreas cromatográficas y los porcentajes utilizados, lo cual muestra que el método desarrollado es viable y confiable para ser aplicado en la cuantificación de metanol en aguas proveniente de diferentes fuentes.
CONCLUSIONES
La medición de metanol en aguas provenientes de diferentes fuentes es posible vía HS-SPME/GC-FID utilizando un tiempo de extracción de 15 minutos a 25 °C, en viales de 15 mL, 5 mL de muestra y temperatura de desorción de 260 °C en un rango de concentraciones entre 0 y 50% de metanol en el agua que se va a analizar. La correlación lineal debe estar cercana a la unidad, los límites de detección y cuantificación bastante bajos (menores de 0,1% de metanol en la mezcla) y bastante altas las precisiones y las exactitudes. La fibra de polidimetilsiloxano es más adecuada para este analito que la fibra de poliacrilato.
Con esta optimización, se abre una nueva posibilidad fácil, confiable y económica en los estudios de sustancias ambientalmente importantes, como en el caso del metanol, comparada con las extracciones en fase líquida, ya que son muchos los residuos obtenidos de esa sustancia en aguas provenientes de diferentes procesos.
AGRADECIMIENTOS
Los autores agradecen a la Universidad de Antioquia y a su programa Estrategia de sostenibilidad de grupos, así como al Ministerio de Agricultura y Desarrollo Rural de Colombia, por el apoyo económico que permitió realizar la experimentación para la presente publicación.
REFERENCIAS BIBLIOGRÁFICAS
1. Araujo, A.; Saraiva, M.; Lima, J.; Korn, M. Flow methodology for methanol determination in biodiesel exploiting membrane-based extraction. Anal Chim Acta. 2008. 613(2): 177 - 183. [ Links ]
2. Mukhopadhyay, S.; Zerella, M.; Alexis, T. A high-yield, liquid-phase approach for the partial oxidation of methane to methanol using SO3 as the oxidant, Advanced Synthesis & Catalysis. 2005. 347 (9): 1203-1206. [ Links ]
3. Pawliszyn, J.; Chen, Y. Solid-Phase Microextraction Field Sampler. Anal. Chim Acta. 2004. 76: 6823-6828. [ Links ]
4. Mahugo, C.; Sosa, Z.; Torres, M.; Santana, J. Methodologies for the extraction of phenolic compounds from environmental samples: New approaches. Molecules 2009. 14: 298-320. [ Links ]
5. Stack, M.; Fitzgerald, G.; O'Connell, S.; James, K. Measurement of trihalomethanes in potable and recreational waters using solid phase micro extraction with gas chromatography-mass spectrometry. Chemosphere. 2000. 41: 1821-1826. [ Links ]
6. Pawliszyn, J. Solid phase microextraction: Theory and practice. New York. Willey-VCH. 1997. pp. 247. [ Links ]
7. Snow, N. Solid-phase micro-extraction of drugs from biological matrices. J. Chromatogr. A. 2000. 885: 445-455. [ Links ]
8. Fritz, J. Analytical solid-phase extraction. Canada, John Wiley & Sons. 1999. pp. 437. [ Links ]
9. Soh, S.; Pauzi, M. Applicability of direct extraction of solid phase micro-extraction to the determination of 54 volatile organic compounds in drinking water. Malaysian Journal Of Chemistry. 2005. 7: 1019-1025. [ Links ]
10. San Juan, P.; Carrillo, J.; Tena, M. Fibre selection base on an overall analytical feature comparison for the solid-phase microextraction of trihalomethanes from drinking water. J Chromatogr. A. 2007. 1139: 27-35. [ Links ]
11. Chao, D.; Kong, S.; Oh, S. Analysis of trihalomethanes in drinking water using headspace-SPME technique with gas chromatography. Water Research. 2003. 37: 402-408. [ Links ]
12. Alonso, A.; Fernández, M.; Tena, M.; Pons, B. Development and validation of a solid-phase microextration method for the analysis of volatile organic compounds in groundwater. Chromatographia. 2003. 57: 369-378. [ Links ]
13. Rianawati, E.; Balasubramanian, R. Optimization and validation of solid phase micro-extraction (SPME) method for analysis of polycyclic aromatic hydrocarbons in rainwater and stormwater. Phys. Chem. Earth. 2009. 34: 857-865. [ Links ]
14. Peña, D. Análisis de datos multivariantes. Madrid. McGraw-Hill, 2002. pp. 539. [ Links ]

