










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804On-line version ISSN 2357-3791
Rev.Colomb.Quim. vol.40 no.2 Bogotá May/Aug. 2011
ESTANDARIZACIÓN DE UN MÉTODO ANALÍTICO PARA EXTRACCIÓN Y CUANTIFICACIÓN DE CIPERMETRINA EN PASTOS
STANDARDIZATION OF ANALYTICAL METHOD FOR EXTRACTION AND QUANTIFICATION OF CYPERMETRHIN IN GRASS
PADRONIZAÇÃO DE MÉTODO ANALÍTICO PARA EXTRAÇÃO E QUANTIFICAÇÃO DE CIPERMETRINA EM PASTAGENS
Yaneth Cardona1,2, Amanda L. Chaparro1,3, Lilia S. Calderón1, Manuel J. Peláez1, Carlos H. García1
1 Departamento de Biología y Química, Facultad de Ciencias Básicas, Universidad de Pamplona, Pamplona, Norte de Santander, Colombia.
2 yanethcar@unipamplona.edu.co
3 achaparro@unipamplona.edu.co
Recibido: 23/05/11 - Aceptado: 09/09/11
RESUMEN
En el presente trabajo se estandarizó un método analítico para la extracción y determinación de cipermetrina en muestras de pastos utilizando microextracción en fase sólida (MEFS) en modo de inmersión con posterior desorción y determinación por cromatografía de gases con detector de ionización de llama (CG-DILL). Las condiciones óptimas para la extracción de cipermetrina en pastos por ID-MEFS fueron: 5 g de muestra se calentaron con 10 mL de solución acuosa de acetona al 1% v/v durante 10 minutos; de esta solución se tomaron 4 mL para llevar a cabo el proceso de extracción por MEFS utilizando una fibra de PDMS con exposición de 30 min a 500 rpm y 60 °C. La desorción del analito se llevó a cabo a 270 °C durante 6 minutos. El procedimiento propuesto mostró comportamiento lineal en el rango probado (5-300 µg/L) con R2 de 0,999. Los límites de detección y cuantificación fueron 1,53 y 4,97 ng/mL, respectivamente, con una desviación estándar relativa de 8,57% (n = 6). El método propuesto en este trabajo permite determinar cipermetrina en muestras de pastos hasta niveles de trazas con una recuperación de 99,08%.
Palabras clave: Pesticida, piretroides, cipermetrina, microextracción en fase sólida (MEFS), cromatografía de gases con detector de ionización de llama (CG-DILL), estandarización
ABSTRACT
In this study, an analytical method has been developed to determine cypermethrin in grass matrices using direct immersion solid-phase microextraction (DI-SPME), coupled to gas chromatography with flame ionization detector (GC-FID). The optimized DI-MEFS experimental procedures to extract cypermethrin in grass matrix were: 5 g of the sample were heated with 10 mL of acetone 1% v/v in water during 10 min. 4 mL of this solution were used for extracting with a polydimethylsiloxane (PDMS)-coated fiber at 60 °C and 500 rpm during 30 min. The analyte desorption was performed at 270 °C for 6 min. The proposed procedure showed linear behavior in the range tested (5-300 mg/L) with R2 of 0.999. The detection and quantification limits were 1.53 and 4.97 ng/ mL respectively with a standard deviation of 8.57% (n = 6). The method proposed in this paper allows determining cypermethrin in samples of grass to trace levels with a recovery of 99.08%.
Key words: Pesticide, pyrethroids, cypermethrin, solid-phase microextraction (SPME), gas chromatography with FID (GC-FID).
RESUMO
No presente estudo, se padronizou um método analítico para a extração e determinação de cipermetrina em amostras de grama usando microextração em fase sólida (MEFS) no modo de imersão com dessorção subseqüentes e determinação por cromatografia gasosa com detector de ionização de chama (CG-DIC). As condições ótimas para a extração de cipermetrina em pastagens por ID-MEFS foram: 5 g de amostra foram aquecidos com 10 mL de solução aquosa de acetona a 1% v/v por 10 minutos. Desta solução, 4 mL foram tomadas para executar o processo de extração MEFS utilizando uma fibra PDMS submetida por 30 min a 500 rpm e 60 °C. A dessorção do analito foi realizada a 270 °C por 6 minutos. O procedimento proposto apresentou um comportamento linear no intervalo testado (500-300 mg/L) com R2 de 0,999. Os limites de detecção e quantificação foram 1,53 e 4,97 ng / mL, respectivamente, com um desvio padrão relativo de 8,57% (n = 6). O método proposto neste trabalho possibilita determinar cipermetrina em amostras de pastagens com uma recuperação de 99,08% nos traços
Palavras-chave: Pesticidas, piretroides, cipermetrina, microextração em fase sólida (MEFS), cromatografia gasosa com FID (CG-DIC).
INTRODUCCIÓN
El incremento en la población mundial requiere mayor cantidad de alimentos; por tanto, el empleo de pesticidas en la agricultura es imprescindible para proteger los cultivos, incrementar el periodo de almacenamiento poscosecha, reducir los costos de producción y disminuir el riesgo de aparición de plagas. Sin embargo, las grandes cantidades de pesticidas que caen en el entorno han causado problemas que afectan el medioambiente y la salud humana, dado que algunos de estos compuestos persistentes se acumulan en la biosfera (aire, agua o suelo) y pueden entrar en la cadena alimentaria de animales, llegando en último término a alcanzar la cadena alimenticia humana, donde varios de ellos se acumulan en algunos órganos vitales y provocan intoxicaciones de distinta gravedad (1).
Actualmente, uno de los pesticidas más usados es la cipermetrina, sintetizado en 1974 e introducido al mercado en 1977. Según la Organización Mundial de la Salud, está clasificado como moderadamente peligroso (clase II); sin embargo, estudios recientes muestran que los efectos de la cipermetrina y de los piretroides en la salud pueden ser más severos de lo que indican evaluaciones toxicológicas previas (2).
La cipermetrina es uno de los insecticidas más usados para controlar una gama amplia de insectos, así como en cosechas, campos agrícolas, invernáculos y tratamiento de poscosecha. En medicina veterinaria se usa para el control de parásitos en animales domésticos y en el ganado. También se utiliza en el hogar para el tratamiento de la sarna y los piojos en los humanos (3).
El control de plagas en praderas de monocultivo, por ejemplo potreros de pasto que sirven de alimentación para el ganado y que son más susceptibles al ataque de dichas plagas, se ha venido realizando últimamente con insecticidas piretroides como la cipermetrina. La aplicación de estos agroquímicos es indiscriminada. Se observa aumento en las dosis y las aplicaciones a frecuencias innecesarias, así como la utilización incorrecta de las formulaciones, lo que convierte estas sustancias en un riesgo potencial para la salud humana, debido a que los residuos de dicho pesticida pueden ingresar por ingestión al animal, dado que consume diariamente más del 10% de su propio peso en pastura, y de allí llegar al organismo humano por la cadena alimenticia (4).
Mientras el número de publicaciones en la determinación de residuos de pesticidas en frutas, hortalizas y otros comestibles es extenso (5-8), la bibliografía dedicada al análisis de pasto (forraje) es relativamente limitada, a pesar de su gran importancia, dado que es el alimento predominante en la dieta alimenticia del ganado vacuno.
Debido a que los plaguicidas se encuentran en niveles de traza en las diferentes muestras medioambientales o biológicas, pues los límites de residuo máximos permitidos (LRM) se expresan en valores de µg/Kg, existe la necesidad de desarrollar un método sensible, fiable, exacto, conveniente, rápido, práctico y selectivo para supervisar las concentraciones de diferentes clases de pesticidas. Para el análisis de residuos de pesticidas en diferentes muestras se han considerado varias técnicas; sin embargo, la mayoría requiere varias horas de análisis y volúmenes grandes de solventes orgánicos, lo cual -además de arriesgar en ocasiones la salud de los operadores- también contribuye a la contaminación medioambiental, puesto que se genera gran cantidad de desechos. Durante la última década, entre las técnicas utilizadas, la microextracción en fase sólida (MEFS) se ha aplicado en la extracción de residuos de diferentes pesticidas piretroides (incluyendo cipermetrina) en diferentes matrices, debido a los beneficios de su uso, tales como simplicidad, tiempo de análisis corto, posibilidad de llevarse a cabo sin el uso de solventes orgánicos (favorece los aspectos de la química verde y protección del ambiente, que se deben tener en cuenta en la valoración de métodos analíticos) y su costo relativamente bajo (la fibra se puede reutilizar hasta más de 100 veces si se le da un uso adecuado).
Dada la importancia de estudiar los residuos de pesticidas por su relación con el bienestar humano y teniendo en cuenta que las investigaciones acerca de estos en pastos es nula, en el presente trabajo se estandarizó un método de extracción y determinación de cipermetrina utilizando las técnicas MEFS y cromatografía de gases con detector de ionización de llama (CG-DILL), que han sido de gran utilidad para el análisis de pesticidas en diversas muestras en los últimos años, pero que hasta donde llega nuestro conocimiento no se han utilizado en el estudio en pastos.
MATERIALES Y MÉTODOSInstrumentos, materiales y reactivos
Para el presente estudio se empleó el estándar analítico cipermetrina (mezcla de isómeros) 95,1% marca Fluka y acetona grado de pureza HPLC marca Merck.
Para las extracciones y los análisis cromatográficos se utilizó un soporte manual para MEFS (Supelco) y cuatro fibras de diferente fase, de las cuales dos eran de polidimetilsiloxano (PDMS) con diferente grosor (100 y 30 µm), una de 65 µm de polidimetilsiloxano-Divinilbenceno (PDMS-DVB) y una de poliacrilato (PA) con un grosor de 85 µm (acondicionadas antes de su uso siguiendo las recomendaciones del fabricante); para la cuantificación de cipermetrina se empleó un cromatógrafo de gases Agilent 6890 con puerto de inyección Split-splitless y detector de ionización de llama (DILL), una columna capilar HP-5 de 60 m X 0,25 mm de diámetro interno x 0,25 µm de espesor.
El gas portador empleado fue helio (99,995%, Aga-Fano) a un flujo de 2 mL/min; la temperatura del inyector a 270 °C (teniendo en cuenta las máximas temperaturas de las fibras utilizadas) y la inyección en modo splitless. El programa de temperatura del horno y las condiciones de desorción de la fibra fueron determinados en este estudio.
Procedimiento y metodología
Todos los análisis realizados se llevaron a cabo por triplicado.
Condiciones cromatográficas
Con el fin de seleccionar las condiciones cromatográficas óptimas para la determinación de cipermetrina, se probaron y modificaron diferentes programaciones del horno utilizadas en diversos estudios para la determinación de los residuos de este y otros piretroides en algunas matrices como frutas y vegetales (9, 10), hojas de tabaco y de té (11), rábano (12). Dichas condiciones se probaron y modificaron con el fin de obtener un tiempo de análisis corto y adecuado para el pesticida de interés en este estudio.
Estandarización del método de extracción
Los experimentos realizados para optimizar los factores que afectan el proceso de extracción fueron llevados a cabo con 3 mL de agua destilada (3,136 ppm de cipermetrina), exponiendo la fibra por inmersión directa a la solución (MEFS-DI) durante 30 min con agitación constante de 300 rpm a temperatura ambiente. Se probaron 4 fibras con recubrimientos diferentes, adición de sal (NaCl) del 0 y 20%, temperaturas de 16, 24, 33, 42, 50, 60 y 70 °C (baño de agua termostáticamente controlado), velocidades de agitación de 0, 100, 300, 500, 700, 900 y 1000 rpm y tiempos de extracción de 10, 20, 30, 40, 50 y 60 minutos, además de volúmenes de muestra de 2, 3 y 4 mL.
Pretratamiento de la muestra
Para el pretratamiento de las muestras de pasto, se compararon cinco tratamientos diferentes (extracción directa al pasto en la solución, infusión, calentamiento, licuado y mezcla licuado-calentamiento) y se observó la eficacia al utilizar agua destilada pura o solución 1% v/v acetona en los diferentes procedimientos. Para el caso de infusión y calentamiento se utilizaron 5 g de pasto con 10 mL (ya sea agua pura o solución acetona), 5 g de muestra con 30 mL en el caso de licuado y mezcla licuado-calentamiento y, por último, al llevar a cabo la extracción directa, 0,5 g de muestra en 4 mL. Todos los experimentos se realizaron por triplicado.
RESULTADOS Y ANALISISCondiciones cromatográficas
Las condiciones cromatográficas óptimas seleccionadas para determinar cipermetrina fueron: temperatura del inyector a 270 °C, inyección modo splitless y flujo del helio a 2 mL/minuto. La programación del horno empieza a 200 °C y se mantiene en esta temperatura por dos minutos; seguidamente aumenta a 20 °C/min hasta 270 °C y de ahí aumenta a 3 °C/min hasta 273 °C, en donde se mantiene constante por un minuto; luego se lleva hasta 278 °C a 0,5 °C/minuto y por último a 10 °C/minuto hasta alcanzar una temperatura final de 290 °C, en la cual se deja por 4 minutos.
Condiciones de extracción
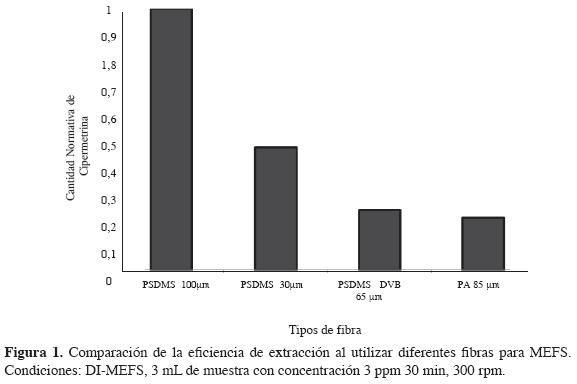
Selección del tipo de fibra. La eficiencia de extracción del método MEFS depende en gran medida de la polaridad del recubrimiento de la fibra, de las propiedades fisicoquímicas del analito designado y de la matriz de la muestra. Los resultados al utilizar las diferentes fibras se observan en la Figura 1.
La polaridad de la fibra puede reforzar la atracción de un analito a un recubrimiento en particular, pero es el espesor de la fibra el que retiene los analitos. La extracción puede lograrse entrampando los analitos en los poros internos o externos de la fibra; así, un analito determina el tipo de recubrimiento que va a usarse. Su selectividad por la fibra se basa principalmente en las diferencias de polaridad y volatilidad (peso molecular) (13,14).
En la Figura 1, se observa que la fibra que mostró mejores resultados para la extracción de la cipermetrina es la PDMS de 100 µm, seguida de la PDMS 30 µm y luego PDMS-DVB 65 µm, sin mucha diferencia con PA 85 µm, que presentó la menor eficiencia de extracción.
Las fibras de PDMS son apolares, lo que las hace aptas para la extracción de pesticidas no polares o de moderada polaridad; por el contrario, la fibra de PA es una fibra más polar y, por tanto, más adecuada en la extracción de compuestos más polares, como fenoles y derivados. En este estudio, la fibra de PA extrajo una pequeña cantidad de cipermetrina muy similar a la extraída por la fibra PDMS/DVB debido posiblemente a su alta afinidad por los aromáticos (13,15).
Las fibras de PDMS/DVB son bipolares y están recomendadas para compuestos volátiles (de bajo peso molecular) como aminas y compuestos nitroaromáticos. En ellas, a diferencia de las fibras de PDMS y PA, la principal interacción entre la fibra y los analitos se produce por adsorción, debido a que es un polímero sólido (15).
La diferencia en la cantidad de cipermetrina extraída por las fibras de diferente grosor de PDMS (100 y 30 µm) se debe a los efectos de tamaño y forma de la molécula del pesticida piretroide. Estos efectos son muy importantes en el proceso de extracción y están relacionados con el grosor del recubrimiento de la fibra. Analitos más pequeños se extraen mejor con recubrimientos gruesos; a medida que se incrementa el tamaño del analito, la eficiencia de extracción de las fibras gruesas disminuye mientras aumenta el de las fibras más delgadas. En un estudio realizado por Robert E. Shirey en 2000 (15), se comparó la eficiencia de extracción de analitos de diferentes tamaños con fibras de diferentes grosores y se encontró que analitos grandes como el DCBP (decachlorobiphenyl), cuyo peso molecular es 499 g/mol, no emigran rápidamente en una fibra de PDMS 100 µm; por consiguiente, la eficiencia de extracción es mayor al utilizar una fibra más delgada como PDMS 30 µm (13).
Para la extracción por MEFS del pesticida piretroide cipermetrina, de carácter no polar, poco volátil, con un peso molecular de 416,3 g/mol, es recomendable usar la fibra de PDMS de 100 µm.
Condiciones de desorción. Dado que la desorción se realiza térmicamente en un cromatógrafo de gases, los parámetros que deben optimizarse son temperatura y tiempo de desorción.
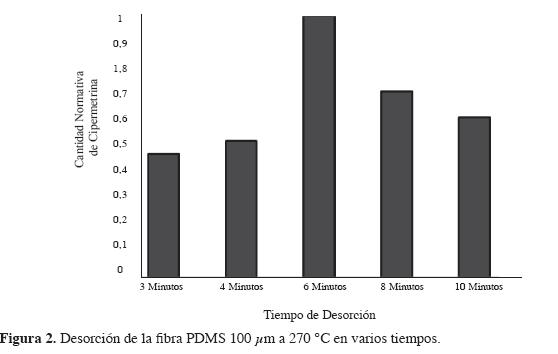
Teniendo en cuenta la temperatura máxima recomendada para el tipo de fibra seleccionada, se fijó la temperatura de desorción (270 °C) y se probaron tiempos de 2, 4, 6, 8 y 10 minutos, realizando una desorción cromatográfica a la fibra después de cada experimento para asegurarse de que en el tiempo escogido salieran de la fibra todos los analitos extraídos, con el fin de prevenir los efectos de memoria en las corridas siguientes. Los resultados se dan a conocer en la Figura 2.
Como se observa en la Figura 2, la cantidad del piretroide analizado aumentó al utilizar tiempos de desorción más prolongados, alcanzando una completa desorción de la cipermetrina a los 6 min, pues antes de este tiempo (2 y 4 minutos) la cantidad de analito presente en la fibra PDMS 100 µm no había sido introducida en su totalidad en la columna analítica por el flujo del gas portador (los blancos de la fibra después de los análisis indicaban restos de cipermetrina) y después de este tiempo (6 minutos) se pierde una cantidad considerable de analito, lo cual puede deberse al largo periodo de exposición de la fibra en el inyector del CG-DILL. Dados los resultados, se determinó realizar la desorción de la fibra PDMS 100 µm en el CG-DILL a 270 °C durante 6 minutos.
Efecto de la adición de sal. Se llevaron a cabo dos pruebas: una sin adición de sal y otra con adición del 20% de NaCl. La adición de sal a muestras acuosas generalmente incrementa la constante de distribución fibra/matriz de moléculas orgánicas neutras. Frecuentemente, para estudios que utilizan EC-MEFS como modo de muestreo, se observan aumentos en la cantidad extraída cuando aumenta la concentración de sal. Sin embargo, las aplicaciones con ID-MEFS son muy pocas, pues al trabajar con disoluciones saturadas en modo directo, se acelera la degradación de la fibra; además, la sal tiene una influencia negativa en la cinética del proceso de ID-MEFS como consecuencia del incremento de la viscosidad y la densidad de la muestra (15,16).
Al adicionar cloruro de sodio (NaCl) se observó una disminución en el rendimiento de extracción, debido a que se modificó la matriz, incrementando la fuerza iónica y reduciéndose la solubilidad del analito, dando como resultado menores recuperaciones. La cantidad extraída con la adición de 20% de NaCl corresponde al 74,85% de la obtenida al llevar a cabo la extracción sin adición de sal.
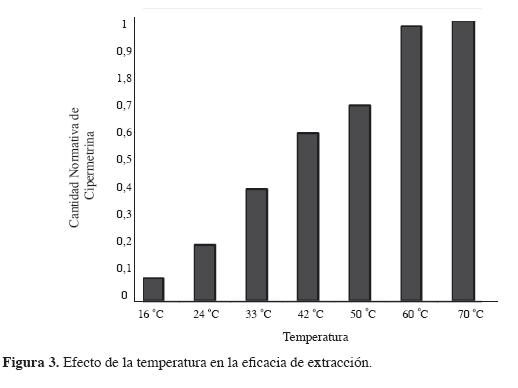
Temperatura de extracción. En MEFS, la temperatura de extracción es uno de los parámetros más importantes, puesto que afecta tanto la sensibilidad como la cinética de extracción. Un incremento en la temperatura de extracción causa un incremento en la proporción de extracción y, simultáneamente, un descenso en la constante de distribución (14,17).
Como se observa en la Figura 3, al aumentar la temperatura de extracción aumenta la cantidad de analito extraído. Dado que la diferencia en la cantidad extraída no es considerable para 60 y 70 °C y debido a que los cromatogramas obtenidos a 70 °C presentan una mala resolución, se determinó trabajar a 60 °C en el proceso de extracción.
Agitación de la muestra en el proceso de extracción. La agitación de la muestra es importante, ya que afecta la cinética y las velocidades del proceso de equilibrio en MEFS (18).
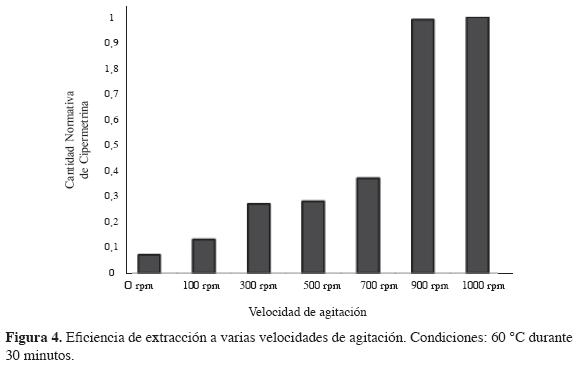
En esta parte del trabajo, se probaron diferentes velocidades de agitación, como se mencionó en la metodología, y se comparó la cantidad de cipermetrina extraída por la fibra en varios tiempos, a dos velocidades de agitación diferentes (500 y 300 rpm), con el fin de mostrar la influencia de la agitación de la muestra en el proceso de extracción.
La respuesta obtenida utilizando el mismo tiempo de extracción es más alta con agitación que sin ella, dado que al agitar la solución se incrementa la difusión de los analitos desde la matriz de la muestra a la fibra y disminuye el tiempo requerido para alcanzar el equilibrio. Los resultados se muestran en la Figura 4, donde se observa que al aumentar la velocidad de agitación, aumenta la cantidad de cipermetrina extraída por la fibra; sin embargo, después de 500 rpm, los cromatogramas no dan muy buena resolución en los picos vecinales y su integración es cada vez más compleja. En esta investigación se utilizó una velocidad de agitación de 500 rpm.
En cuanto a los resultados obtenidos en la comparación de la cantidad de cipermetrina extraída por la fibra en varios tiempos, a dos agitaciones diferentes (Figura 5), la agitación tiene una influencia positiva en la extracción, pues el tiempo de equilibrio disminuye progresivamente con el incremento de las rpm, a tal punto que la cantidad obtenida de cipermetrina en 60 minutos a 300 rpm se puede extraer a 500 rpm en 40 minutos. Sin embargo, con esta velocidad de agitación, el tiempo de equilibrio sigue siendo mayor de una hora.
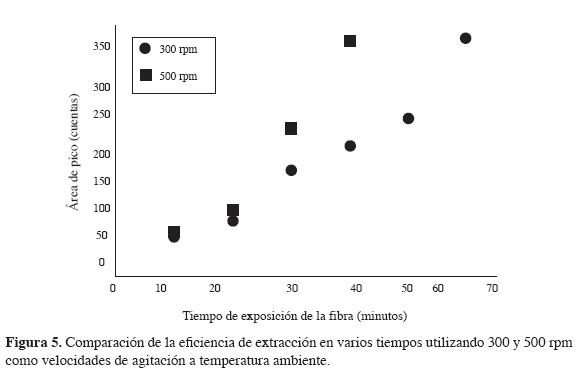
Tiempo de extracción. Al desarrollar un método analítico basado en MEFS se busca determinar el tiempo necesario para llegar al estado de equilibrio característico de cada analito-fibra e intentar trabajar en esas condiciones. En esta etapa, la mayoría de los parámetros fundamentales han sido determinados para las condiciones óptimas de MEFS. El tiempo de extracción es definido como el tiempo a partir del cual la cantidad de analito extraída por la fibra se mantiene constante; en MEFS es un parámetro muy importante para tener en cuenta. Generalmente, la cantidad extraída se incrementa rápidamente en los primeros 5 minutos, pero el equilibrio no es alcanzado hasta mucho después. En este estudio, se probaron tiempos de 10, 20, 30, 40, 50 y 60 minutos con las condiciones óptimas seleccionadas hasta el momento.
Para algunos compuestos, el tiempo necesario para alcanzar el equilibrio es muy elevado; por consiguiente, se opta por trabajar en condiciones de no equilibrio y se seleccionan tiempos de extracción inferiores para no alargar el tiempo de análisis. En estos casos es muy importante controlar estrictamente el tiempo de extracción, ya que pequeñas oscilaciones en la medida del tiempo pueden variar de forma considerable la cantidad de analito extraída (19).
Generalmente, en extracción directa (DI-MEFS) de muestras acuosas puras con agitación magnética, utilizando una fibra de PDMS de 100 µm, los tiempos de equilibrio son menores de una hora para compuestos que tienen constantes de distribución estimadas en menos de 10.000. Los coeficientes de partición de los analitos entre el PDMS y el agua (KPDMS/w) son proporcionales a sus coeficientes de partición octanol-agua (Ko/w); por tanto, teniendo en cuenta el Ko/w de la cipermetrina, su KPDMS/w es mayor de 100.000, así que el tiempo necesario para alcanzar el equilibrio entre la solución y la fibra de PDMS 100 µm es posiblemente superior a una hora (20).
Experimentalmente, se observó que al aumentar el tiempo de exposición de la fibra aumentó la cantidad de cipermetrina extraída; sin embargo, en 60 minutos de exposición la fibra no alcanzó el equilibrio. Así, en esta investigación se trabajó en condiciones de no equilibrio con 30 minutos, lo cual requiere control estricto de las variables temperatura, tiempo y agitación constante en todos los análisis.
Influencia del volumen y tipo de muestreo. El volumen de muestra es otro parámetro para tener en cuenta al desarrollar un método analítico basado en MEFS.
Una vez escogidas las condiciones óptimas de los diferentes parámetros, se estudió la influencia de la cantidad de muestra, observando la diferencia al usar 2, 3 y 4 mL en el modo ID-MEFS, y se compararon los resultados obtenidos en las mismas condiciones por el muestreo EC-MEFS con 2 mL de muestra.
En la primera parte, correspondiente a la comparación de la cantidad extraída de cipermetrina por la fibra en modo ID-MEFS a diferentes volúmenes, se observó que a medida que el volumen de la muestra se incrementó, la cantidad de analito absorbida por la fibra aumentó; en consecuencia, se decidió utilizar 4 mL de muestra.
En esta segunda parte, correspondiente a la comparación de los dos modos de extracción (ID-MEFS y EC-MEFS), los resultados muestran que al realizar la extracción de cipermetrina, la cantidad obtenida por inmersión es mayor que la obtenida por espacio cabeza. La respuesta obtenida por EC-MEFS (con las mismas condiciones que ID-MEFS y estrictamente controladas) fue 29,7% de lo que se obtiene con el modo ID-MEFS.
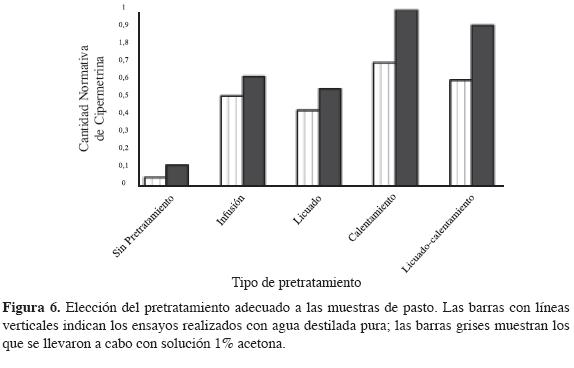
Pretratamiento de las muestras vegetales. En la Figura 6 se pueden observar los resultados obtenidos al utilizar los diferentes métodos propuestos (infusión, calentamiento, licuado, mezcla licuado-calentamiento y sin pretratamiento del pasto).
Como se observa en la Figura 6, los mejores resultados se obtienen al utilizar calentamiento de la muestra vegetal utilizando solución 1% acetona; los resultados menos satisfactorios se debieron a no realizar ningún tipo de pretratamiento antes del proceso de extracción utilizando agua destilada. La cantidad extraída de esta forma es el 7% de la cantidad obtenida en la extracción al realizar el pretratamiento del pasto por calentamiento con 6 mL de solución 1%.
VALIDACIÓN DEL MÉTODO PROPUESTO
Las condiciones óptimas para la extracción y la determinación de cipermetrina en muestras de tejido vegetal por el método DI-MEFS-CG-DILL son: 5 g de pasto son sometidos a calentamiento con 10 mL de solución 1% acetona durante 10 minutos; 4 de estos mL son analizados con MEFS durante 30 min con agitación constante a 500 rpm y a una temperatura de 60 °C sin adición de sal.
Este método fue validado para análisis de cipermetrina en muestras de pasto evaluando la linealidad, el límite de detección, el límite de cuantificación, la precisión y la exactitud.
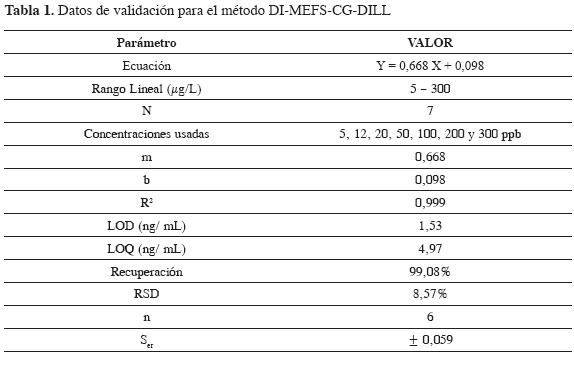
Para evaluar el desempeño del procedimiento DI-MEFS-CG-DILL, los parámetros fueron medidos usando muestras de pasto provenientes del municipio de Pamplona (ubicado en la zona suroccidental del departamento Norte de Santander -Colombia) con concentraciones conocidas del piretroide investigado. Los parámetros de validación son presentados en la Tabla 1.
Linealidad, límite de detección (LOD) y límite de cuantificación (LOQ)
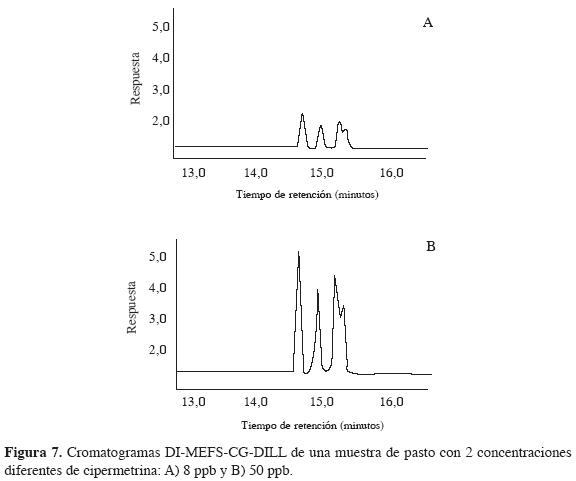
Dos cromatogramas obtenidos en la determinación de cipermetrina a 2 concentraciones diferentes (8 y 50 ppb) son presentados en la Figura 7.
La linealidad del método fue evaluada utilizando 7 concentraciones diferentes en el rango 5 - 300 ng/ mL obteniendo un coeficiente de correlación (R2) que exhibe una buena linealidad en el rango de concentraciones examinadas (Tabla 1). Cada análisis fue hecho por triplicado.
Los valores calculados de LOD (proporción señal a ruido de 3; S/N = 3) y LOQ (proporción señal a ruido de 10; S/N = 10) se muestran en la Tabla 1.
Exactitud, precisión y reproducibilidad. La evaluación de la exactitud del método se basó en la recuperación de cantidades conocidas de cipermetrina en muestras con tres concentraciones diferentes (5 ppb, 50,6 ppb y 200,13 ppb), las cuales fueron analizadas por triplicado de acuerdo a las condiciones óptimas. La precisión del método fue evaluada durante un día y los valores de repetibilidad fueron calculados analizando muestras (n = 6) a una concentración de 5 µg/L.
Los valores obtenidos de recuperación de cipermetrina y desviación estándar relativa (RSD) (Tabla 1) se consideran adecuados para el análisis de residuos de pesticidas y muestran que la reproducibilidad de este método es satisfactoria. Por consiguiente, utilizarse en el análisis de residuos de cipermetrina en muestras de pasto.
CONCLUSIONES
La metodología utilizada para la extracción y cuantificación de cipermetrina en pastos validada en este trabajo, cumple con los parámetros exigidos para la validación de un método analítico, garantizando de esta manera que los resultados obtenidos al aplicarlo en el análisis de muestras de pasto son confiables.
Con este método analítico se agiliza el proceso de análisis, disminuyendo considerablemente la generación de desechos tóxicos y las cantidades de reactivos utilizados, proporcionando validez y confiabilidad en los resultados de cuantificación.
AGRADECIMIENTOS
Los autores agradecen al Ministerio de Agricultura y Desarrollo Rural, de la República de Colombia, por su apoyo financiero a esta investigación bajo el macroproyecto titulado Mejoramiento de la Agroindustria Cárnica con relación a la Presencia de Sustancias Xenobióticas en los Subsistemas Agrícola y Pecuario, código MDR 2008H2468-3968.
REFERENCIAS BIBLIOGRÁFICAS
1. Esteve Turrillas, F.A. Preparación de muestras para el análisis de plaguicidas mediante microondas y fluidos presurizados. Tesis doctoral. Universitat De Valencia. Departamento de Química Analítica. Valencia, España. 2007. 215 p. [ Links ]
2. Jiménez, L.; Quilodrán, J.; Miranda, J.P.; Rodríguez, H. Efecto de dosis única intraperitoneal de cipermetrina en la corteza cerebral somatosensorial de ratones. International J. Morphology. 2008. 26: 19-26. [ Links ]
3. Fortin, M.C.; Bouchard, M.; Carrier, G.; Dumas, P. Biological monitoring of exposure to pyrethrins and pyrethroids in a metropolitan population of the Province of Quebec, Canada. Enviro Research. 2008. 107: 343-350. [ Links ]
4. Loaiza, A.C. Lineamientos de políticas sobre uso y manejo mesurado de plaguicidas con énfasis en el sector agropecuario y forestal del departamento de Antioquia. Convenio Interinstitucional, 23 de diciembre de 2003 entre Ceiba, Cornare y Dama-Antioquia. Medellín Colombia. 2005. 142 p. [ Links ]
5. Garrido, A.; Plaza-Bolaños, P.; Martínez, J.L. Comparison of tandem-in-space and tandem-in-time mass spectrometry in gas chromatography determination of pesticides: Application to simple and complex food samples. J. Chromatography A. 2008. 1203: 229-238. [ Links ]
6. Cortés, J.M.; Vázquez, A.; Santa-María, G.; Blanch, P. Pesticide residue analysis by RPLC-GC in lycopene and other carotenoids obtained from tomatoes by supercritical fluid extraction. Food Chem. 2009. 113: 280-284. [ Links ]
7. Gilbert López, B.; García-Reyes, J.F. ; Molina-Díaz, A. Sample treatment and determination of pesticide residues in fatty vegetable matrices: A review. Talanta. 2009. 79: 109-128. [ Links ]
8. Pizzutti, I.R.; Vreuls, R.J.; de Kok, A.; Roehrs, R.; Martel, S.; Friggi, C.A.; Zanella, R. Design of a compressed air modulator to be used in comprehensive multidimensional gas chromatography and its application in the determination of pesticide residues in grapes. J. Chromatography A. 2009. 1216: 3305-3311. [ Links ]
9. Beltrán, J.; Peruga, A.; Pitarch, E.; López, F.J.; Hernández, F. Application of solid-phase microextraction for the determination of pyrethroid residues in vegetable samples by GC-MS. Anal Bioanal Chem. 2003. 376: 502-511. [ Links ]
10. Zawiyah, S.; Che Man, Y.B.; Nazimah, S.A.H.; Chin, C.K.; Tsukamoto, I.; Hamanyza, A.H.; Norhaizan, I. Determination of organochlorine and pyrethroid pesticides in fruit and vegetables using SAX/PSA clean-up column. Food Chem. 2007. 102: 98-103. [ Links ]
11. Hou, Y.; Cao, Q.; Xie, X.; Wanjg, B.; Xu, J.; Yang, Y. Determination of pyrethroid pesticide residues in tobacco leaves and tea using stir bar sorptive extraction-thermal desorption and gas chromatography-mass spectrometry. Chinese J. Chromatography. 2007. 25: 25-29. [ Links ]
12. Nguyen,T.D.; Han, E.M.; Seo, M.S.; Kim, S.R.; Yun, M.Y.; Lee, D.M.; Lee., G.H. A multi-residue method for the determination of 203 pesticides in rice paddies using gas chromatography/mass spectrometry. Anal chim A. 2008. 619: 67-74. [ Links ]
13. Shirey, R.E. Optimization of extraction conditions and fiber selection for semivolatile analytes using Solid-Phase Microextraction. J. Chromatographic Science. 2000. 38: 279-288. [ Links ]
14. Pawliszyn, J. 2002. Sampling and simple preparation for field and laboratory. XXXVII. Editado por D. Barceló. Elsevier Science BV. Wilson & Wilson's. Comprehensive Analytical Chemistry. 1131 p. [ Links ]
15. Fernández Torroba, M.Á. Estudio y modelización de la contaminación del acuífero del río Najerilla. Aplicación de la microextracción en fase sólida para la determinación de los contaminantes. Tesis Doctoral. Universidad de la Rioja. Departamento de Química. 2004. 391 p. [ Links ]
16. Fernández-Álvarez, M.; Llompart, M.; Lamas, J.P.; Lores, M.; García-Jares, C.; Cela, R.; Dagnac, T. Simultaneous determination of traces of pyrethroids, organochlorines and other main plant protection agents in agricultural soils by headspace solid-phase microextraction-gas chromatography. J. Chromatography A. 2008. 1188: 154-163. [ Links ]
17. Pawliszyn, J. Theory of solid-phase microextraction. J. Chromatographic Science. 2000. 38: 270-278. [ Links ]
18. Beceiro-González, E.; Concha-Graña, E.; Guimaraes, A.; Goncalves, C.; Muniatequi-Lorenzo, S.; Alpendurada, M.F. Optimisation and validation of a solid-phase microextraction method for simultaneous determination of different types of pesticides in water by gas chromatography-mass spectrometry. J. Chromatography A. Febrero, 2007. 1141(2): 165-173. [ Links ]
19. Gianelli Barra, M.P. Interacción entre compuestos aromáticos del jamón curado y compuestos proteicos musculares solubles. Tesis Doctoral. Valencia, España.: Universitat de Valencia. Departamento de Medicina Preventiva, Salud Pública, Bromatología, Toxicología y Medicina Legal. 2004. 230 p. [ Links ]
20. Cháfer Pericás, C. Acoplamiento microextracción en fase sólida-cromatografía. Tesis Doctoral. Valencia, España: Universitat de Valencia. Departamento de Química Analítica. 2006. 372 p. [ Links ]

