










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804On-line version ISSN 2357-3791
Rev.Colomb.Quim. vol.40 no.2 Bogotá May/Aug. 2011
ANÁLISIS DE RESIDUOS DE PLAGUICIDAS EN TOMATE MEDIANTE EL USO DE QuEChERS Y CROMATOGRAFÍA LÍQUIDA ULTRARRÁPIDA ACOPLADA A ESPECTROMETRÍA DE MASAS
ANALYSIS OF PESTICIDE RESIDUES IN TOMATO USING QuEChERS SAMPLE PREPARATION AND ULTRA FAST LIQUID CHROMATOGRAPHY COUPLED TO MASS SPECTROMETRY
ANÁLISE DE RESÍDUOS DE PESTICIDAS EM TOMATE USANDO QuEChERS E CROMATOGRAFIA LÍQUIDA ULTRA RÁPIDA ASSOCIADA Á ESPECTROMETRIA DE MASSAS
Diego A. Ahumada1,2, Adriana M. Zamudio1
1 Laboratorio de Inocuidad Química. Centro de Biosistemas. Facultad de Ciencias Naturales e Ingeniería. Universidad Jorge Tadeo Lozano. Bogotá D.C., Colombia.
2 diego.ahumadaf@utadeo.edu.co
Recibido: 23/07/11- Aceptado: 09/09/11
RESUMEN
Se desarrolló un método multirresiduo para la determinación de 24 plaguicidas de diferentes características fisicoquímicas en tomate. El proceso de extracción se basó en el método QuEChERS y la determinación de los compuestos se realizó mediante cromatografía líquida ultrarrápida acoplada a espectrometría de masas. Los resultados de la validación indicaron que el método presenta porcentajes de recuperación de entre el 71,3 y el 114%, coeficientes de variación inferiores al 20%, límites de detección entre 0,002 y 0,225 mg/kg, límites de cuantificación entre 0,009 y 0,750 mg/kg. En los estudios de linealidad, para todos los parámetros evaluados, se obtuvieron resultados adecuados en los rangos de trabajo. Para probar la metodología desarrollada, se evaluaron muestras de frutos de tomate provenientes del departamento de Boyacá y se halló indoxacarb en cantidades inferiores al límite máximo de residuos.
Palabras clave: plaguicidas, espectrometría de masas, cromatografía líquida.
ABSTRACT
We developed a multiresidue method for determining 24 pesticides of different physicochemical characteristics presents on tomato. The extraction process is based on the QuEChERS method, and the determination of the compounds was performed using ultra performance liquid chromatography coupled to mass spectrometry. The validation results indicated that the method presents recoveries between 71.3 and 114%, coefficients of variation below 20%, detection limits from 0.002 to 0.225 mg/kg, quantification limits between 0.009 and 0.750 mg/kg. In linearity studies, the results were adequate in the ranges evaluated for all the parameters in this study. Finally, to test methodology, some samples from the Department of Boyacá were analyzed. Indoxacarb was present in amounts below the maximum residues limit.
Key words: Pesticides, mass spectrometry, liquid chromatography.
RESUMO
Desenvolvemos um método multiresíduo para determinar 24 pesticidas de diferentes características físico-químicas em tomate. O processo de extração é baseado no método QuEChERS. Na determinação dos compostos utilizou-se cromatografia líquida de ultra-desempenho acoplada a espectrometria de massa. Os resultados da validação indicaram que o método apresentou recuperações entre 71,3 e 114%, os coeficientes de variação estiveram abaixo de 20%, os limites de detecção entre 0,002 e 0,225 mg/kg, os limites de quantificação entre 0,009 e 0,750 mg/kg. Em estudos de linearidade, se descobriu que, para todos os parâmetros, os resultados foram adequados. Para testar a metodologia desenvolvida, testaram amostras do departamento de Boyacá e constatou a presença de indoxacarbe em quantidades inferiores ao limite máximo de resíduos.
Palavras-chave: pesticidas, espectrometria de massas.
INTRODUCCIÓN
En los últimos años, el análisis de residuos de plaguicidas es un campo de la química analítica que ha despertado un gran interés en diferentes centros de investigación, universidades y organizaciones gubernamentales, consecuencia del gran número de nuevos compuestos que se introducen en el mercado, las nuevas regulaciones establecidas -pues estas resultan ser cada día más restrictivas en cuanto al límite máximo de residuos (LMR)- y el interés social, económico y académico por mejorar la calidad en el consumo de los alimentos, lo cual tiene un gran impacto en el mercado mundial (1).
Como consecuencia de las características particulares de los plaguicidas y la necesidad de contar con métodos cada vez más precisos, específicos, exactos, rápidos y robustos, las técnicas cromatográficas resultan ser la opción más adecuada para este tipo de determinaciones cualitativas y/o cuantitativas (2). La cromatografía es probablemente la técnica analítica más empleada en laboratorios debido a numerosas razones: i) los sensibles métodos de detección existentes, ii) la capacidad de separar, identificar y analizar de manera cualitativa y cuantitativa, y iii) la alta rapidez de los análisis cromatográficos, los cuales van desde varios minutos hasta algunos segundos en ciertas circunstancias (3).
A principios de la última década, la cromatografía de alta eficiencia acoplada a espectrometría de masas se ha convertido en la técnica por excelencia para la determinación de residuos de contaminantes (4-6). Esta preferencia se debe a la enorme diversidad de propiedades fisicoquímicas que presentan estos compuestos, sumada a la gran versatilidad y a la mayor cantidad de grupos químicos que pueden analizarse, comparada con la cromatografía de gases (7).
Los desarrollos tecnológicos actuales referentes a cuadrupolos, trampas iónicas y otros filtros de masas proveen a los sistemas de espectrometría de masas de una alta especificidad y selectividad en un amplio rango de masas, lo cual ha conducido a la eliminación casi completa de interferentes en el registro de la señal de los analitos, manteniendo a su vez una adecuada sensibilidad. Estos desarrollos han marcado una tendencia a la evolución de técnicas analíticas que requieran poca preparación de muestra y presenten una mínima retención cromatográfica (8, 9).
En 2003 buscando superar todos los inconvenientes prácticos de los métodos multirresiduo existentes de la época y aprovechando la selectividad de los espectrómetros de masas, se introdujo una nueva metodología de análisis de residuos de plaguicidas en material vegetal, denominada QuEChERS (acrónimo en inglés de Quick, Easy, Cheap, Effective, Rugged, y Safe) (10). Este método implica dos etapas fundamentales: en la primera, los plaguicidas se extraen con acetonitrilo en presencia de sulfato de magnesio y acetato de sodio. La segunda etapa corresponde a una limpieza del extracto con base en la aplicación de extracción en fase sólida por dispersión con una amina primaria secundaria (PSA). En la actualidad se realizan diferentes modificaciones al método, de manera que continuamente se logran adaptaciones a nuevas matrices o compuestos de diferentes características (11-13). Así, junto con la cromatografía líquida acoplada a espectrometría de masas, este método facilita el análisis de un número elevado de compuestos en un tiempo muy corto.
En este artículo aparecen los resultados de la validación de la metodología analítica desarrollada (14), basada en el método QuECHERS, para la determinación de residuos de plaguicidas en tomate (Solanum lycopersicum), mediante cromatografía líquida ultrarrápida acoplada a espectrometría de masas (UFLC-MS, de sus siglas en inglés). Asimismo, se muestran los resultados en la evaluación de muestras provenientes de un cultivo de tomate ubicado en el departamento de Boyacá (Colombia).
MATERIALES Y MÉTODOS
Materiales de referencia, reactivos y soluciones
Se trabajó con estándares de plaguicidas de pureza mayor al 95%, proveniente de las casas comerciales Dr Ehrenstorfer y Chemservice. Las soluciones madre fueron preparadas en concentraciones cercanas a 500 µg/mL en acetonitrilo o metanol y almacenadas en frascos ámbar a -20 °C. La mezcla de plaguicidas se preparó en metanol, tomando diferentes volúmenes de cada una de las soluciones madre hasta obtener un rango de concentraciones entre 0,64 y 9,95 µg/mL. Esta solución se almacenó en frasco ámbar a -20 °C. Los solventes empleados en este estudio fueron J.T. Baker grado HPLC. Para los ensayos de extracción se utilizaron sales de QuEChERS Restek Q-Sep TM y para la limpieza de los extractos se emplearon los adsorbentes Restek dSPE Q-Sep TM. Se empleó como estándar subrrogado trifenilfosfato (TPP) y como estándar interno (E.I.) fosfato de tris-(1,3-dicloroisopropil).
Instrumentos y equipos
El análisis cromatográfico se llevó a cabo en un cromatógrafo líquido ultrarrápido Shimadzu Prominence (Maryland, CA, EUA), acoplado a un detector selectivo de masas LCMS-2020. Se empleó un generador de nitrógeno ABN2ZE Peak Scientific (Billerica, USA). El cromatógrafo cuenta con un inyector automático SIL20A UFLC 7673 Shimadzu (Maryland, CA, EUA), una bomba binaria de alta presión, un sistema de desgasificación en línea y un horno para el control de la temperatura de la columna. El control del sistema cromatográfico y la adquisición y el procesamiento de datos se realizaron con el software Lab Solutions versión 3.5.
Condiciones cromatográficas
Los análisis se realizaron en una columna Shim Pack (6 cm x 2 mm d.i., tamaño de partícula de 2,1 Μm y fase estacionaria C18), en modo de gradiente lineal con ácido fórmico al 0,1% (P/V) y acetato de amonio 5 mM en agua Milli-Q (A). La fase orgánica empleada fue acetonitrilo (B). El programa de elución utilizado, expresado como porcentaje de B, inicia a 0% (0 min) aumenta a 20% en 0,01 min, se mantiene por 0,3 min, posteriormente llega hasta 55% en 8,5 min y luego aumenta hasta el 100% en los siguientes 1,5 min. Finalmente, se mantiene por 1,5 min. Para restituir la fase móvil a la condición inicial del análisis se pasa de 100% a 0% de B en 2 min, donde se mantiene por 5 min para equilibrar la columna. El tiempo total de análisis fue 12 min. El volumen de inyección fue 5 µL, la temperatura de columna 45 °C y el flujo de fase móvil 0,3 mL/min.
Condiciones de la interfaz y espectrómetro de masas
El equipo cuenta con una interfaz tipo DUIS (ESI, APCI) que se operó en modo ESI, con un flujo de gas de secado de 15 L/min y un flujo de gas nebulizador de 1,5 L/min. Las temperaturas del bloque de calentamiento y de la línea de eliminación del solvente correspondieron a 200 y 250 °C, respectivamente.
Los análisis fueron llevados de manera simultánea en modo positivo y negativo, el voltaje aplicado en el capilar correspondió a 4500 y -4500 V, respectivamente. Todos los análisis fueron realizados en modo SIM. La Tabla 1 muestra los iones seleccionados y el modo de ionización para cada uno de los compuestos.

Método de extracción
Para el desarrollo y validación de la metodología, se utilizaron muestras de tomate (Solanum lycopersicum) certificadas como productos orgánicos, obtenidas en supermercados de cadena. Se lavaron con abundante agua y se retiró todo material ajeno a la fruta que pudiera encontrarse en su superficie. Para asegurar que las muestras se encontraran libres de plaguicidas se evaluaron mediante UFLC-MS.
Para la extracción de los plaguicidas se empleó el método QuEChERS con algunas modificaciones (14). Para realizar este proceso, en un tubo de centrifuga se pesaron 10 g de muestra homogeneizada, se adicionó TPP, se dejó en reposo por 10 min y, transcurrido ese tiempo, se adicionaron 10 mL de acetonitrilo acidificado con ácido acético al 1% (V/V), 4 g de MgSO4 anhidro y 1 g de AcONa, agitado manualmente por 1 min. Posteriormente, se llevó al baño de ultrasonido por 10 min, se centrifugó a 4500 rpm por 5 min y, con ayuda de una pipeta, se tomaron 10 mL del sobrenadante (Solución A), los cuales se transfirieron a un tubo de centrifuga de 15 mL.
Para el proceso de limpieza, por cada mililitro de extracto (solución A) se adicionaron 25 mg de PSA (amina primaria/secundaria) y 150 mg de MgSO4 anhidro, se agitó manualmente por 30 s y se centrifugó por 2 min a 4500 rpm. Finalmente, el sobrenadante se filtró a través de una membrana de 0,22 Μm de PTFE (Solución B), se transfirió a un vial de cromatografía donde se adicionó el estándar interno (E.I.).
Validación de la metodología
Se evaluó la especificidad de la metodología mediante la comparación de blancos de matriz y blancos de matriz fortificados; además, para evaluar la exactitud y la precisión se fortificaron muestras de tomate a tres diferentes concentraciones equivalentes a los niveles 1, 3 y 5 de la curva de calibración (Tabla 2). Para evaluar la precisión intermedia se analizaron muestras fortificadas a los niveles 1,3 y 5 en diferentes periodos de tiempo.
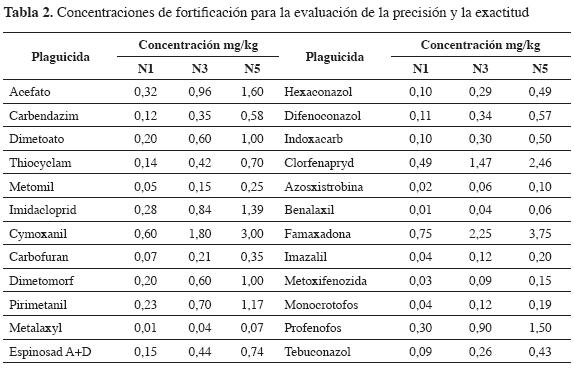
La evaluación de la linealidad se realizó preparando curvas de calibración en blancos de matriz; cada curva está compuesta de cinco puntos o niveles de concentración. La evaluación de los límites de detección y cuantificación se realizó siguiendo método de la IUPAC (15). Se inyectaron blancos de tomate y se evaluó el ruido en la zona donde se encontraba cada uno de los plaguicidas; posteriormente, se fortificaron muestras a los límites de cuantificación y se realizó la extracción para determinar la precisión y la exactitud del método. Finalmente, para evaluar la robustez del método se empleó el modelo de Youden Steiner (16). Todos los ensayos se realizaron por quintuplicado y los resultados obtenidos fueron analizados de acuerdo con el parámetro evaluado, mediante el paquete estadístico PASW STATISTICS 18 del entorno IBM® SPSS®
Evaluación de muestras reales
El método se aplicó a muestras reales tomando muestras en una finca de producción de tomate Bachué ubicada en la vereda de Sopotá, municipio de Villa de Leyva del departamento de Boyacá (N 5° 37' 24,4 y O 73° 33' 3,03). Esta finca se encuentra a una altura de 2094 msnm; la producción se realiza bajo invernadero.
Durante la época de cosecha, se realizaron cuatro muestreos durante un mes. Semanalmente se seleccionaron al azar frutos de tomate que se encontraran en el punto de maduración establecido por el productor. Cada muestra estuvo compuesta por 1 kg de tomates aproximadamente. Las muestras fueron transportadas inmediatamente al laboratorio y se procedió a realizar su análisis por duplicado. De esta manera, durante el periodo de evaluación se analizaron 16 muestras.
RESULTADOS Y DISCUSIÓN
Especificidad
La comparación entre el blanco de matriz obtenido y el blanco fortificado (concentración más baja de la curva de calibración) mostró que cumple con los parámetros que establece la guía de validación de métodos analíticos de la Unión Europea (17), ya que ninguno de los compuestos evaluados presentó interferencias superiores al 30% de la respuesta. Sin embargo, para algunos compuestos, como el caso de carbendazim y difenoconazol, se encontraron algunas interferencias que dificultaron el proceso de integración; por tanto, se decidió aumentar la concentración de estos compuestos para evitar este tipo de inconvenientes en la cuantificación.
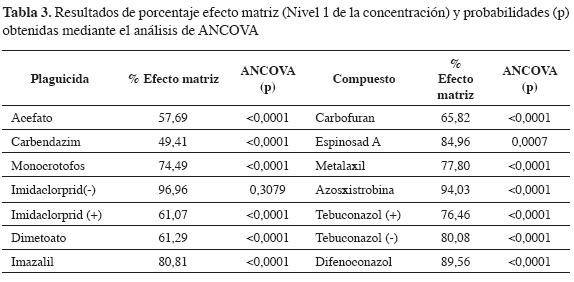
Efecto matriz
En el análisis de residuos de plaguicidas, el efecto matriz se define como la variación de la respuesta cromatográfica inducida por los compuestos de la matriz (18). Para el caso específico de la cromatografía líquida acoplada a espectrometría de masas, se ha encontrado que el proceso de ionización que ocurre en la interfaz del instrumento se ve afectado por compuestos que coeluyen con los analitos que se desea determinar (19). De esta manera, se evaluó el efecto matriz mediante la comparación de curvas de calibración realizadas en solvente y en blanco de matriz. Los resultados de estos ensayos se evaluaron mediante el cálculo del porcentaje de efecto matriz (relación áreas matriz-solvente) y análisis de covarianza (18). En la Tabla 3 se observan algunos de los resultados de esta evaluación: la mayoría de los compuestos presentan porcentajes de efecto matriz inferiores al 100%, lo cual indica que el efecto predominante de los componentes de la matriz es el de la supresión de la ionización de los plaguicidas analizados. Al realizar el análisis estadístico (ANCOVA) se concluye que en todos los compuestos se presenta efecto matriz (p < 0,05), a excepción de imidaclorprid en modo negativo, lo cual es objeto de otro estudio (20).
Linealidad
De acuerdo con los resultados de efecto matriz obtenidos, la linealidad de la metodología fue evaluada mediante la preparación de las curvas de calibración en blanco de matriz (21,22). La Tabla 4 muestra los rangos de concentración evaluados y los parámetros de regresión (pendiente, intercepto y coeficiente de correlación).
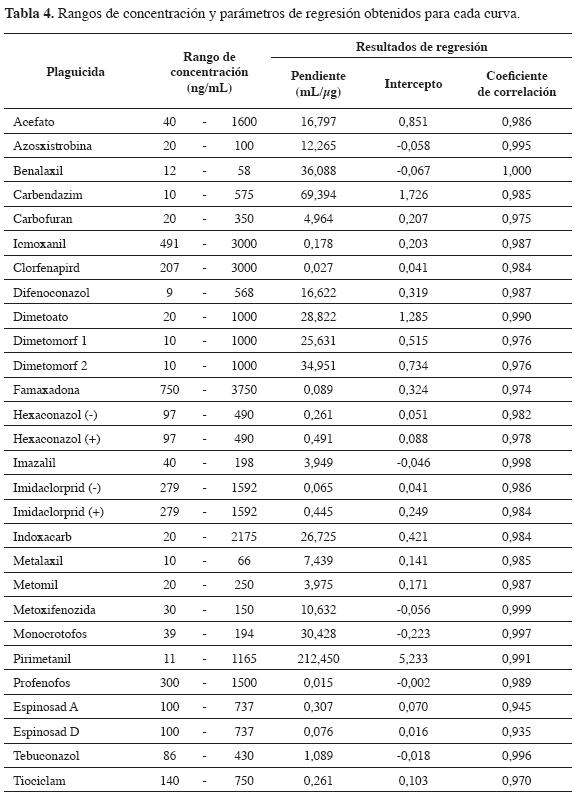
El primer aspecto que se evaluó de la linealidad fue la significancia de la regresión lineal. Mediante una análisis de varianza se estableció la validez del modelo y = mx + b con a = 0,05; por consiguiente, las curvas de calibración se ajustan efectivamente al modelo planteado. Una vez se estableció la significancia del modelo, se determinó si los parámetros m y b son estadísticamente iguales a cero, mediante una prueba t. Se encontró que, para todos los compuestos, la pendiente es estadísticamente diferente de cero (a = 0,05); excepto, para espinosad (D) e imidacloprid (-), en que el intercepto es estadísticamente igual a cero (a = 0,05), lo cual implica que para los compuestos restantes es indispensable emplear el intercepto en el momento de la cuantificación. Otro aspecto evaluado corresponde a la correlación entre las variables X y Y, (concentración y respuesta cromatográfica, respectivamente). Para esta evaluación se aplicó una prueba t, la cual indicó que todos los coeficientes de correlación son estadísticamente iguales a 1(a = 0,05). De igual manera, se realizó una prueba de falta de ajuste o desvío de linealidad cuyo objetivo es evaluar los rangos de concentración en los cuales se elaboró la curva y determinar si se presentan desvíos a concentraciones altas o bajas de la respuesta en función de la concentración. Esta prueba reveló que ningún compuesto presentó desvíos de linealidad (a = 0,05). Finalmente, de acuerdo con la guía de validación de la Unión Europea, se determinaron los residuales y se halló que ninguno excede el 20% (17).
Precisión
La precisión de la metodología para cada uno de los compuestos se evaluó con estudios de repetibilidad y de precisión intermedia (interdías), aplicando la determinación del coeficiente de variación (%CV).
La Tabla 5 presenta los %CV de cada uno de los compuestos evaluados. Para repetibilidad, se obtuvieron %CV inferiores del 15%. Compuestos como carbofuran, carbendazim, clorfenapird, hexaconazol, entre otros, presentan valores inferiores del 5% en algunas ocasiones, lo cual indica que la dispersión es bastante baja. En cuanto a los valores obtenidos para precisión intermedia, se observa un aumento considerable de la dispersión; por ejemplo, en el caso de acefato en el nivel 5, se pasa de un %CV de 7,4 a 17,6%. Estas variaciones se pueden atribuir a fenómenos de dispersión y perturbaciones en las respuestas cromatográficas, teniendo en cuenta que la metodología analítica no implica mayores variaciones de acuerdo con lo publicado en literatura (17).
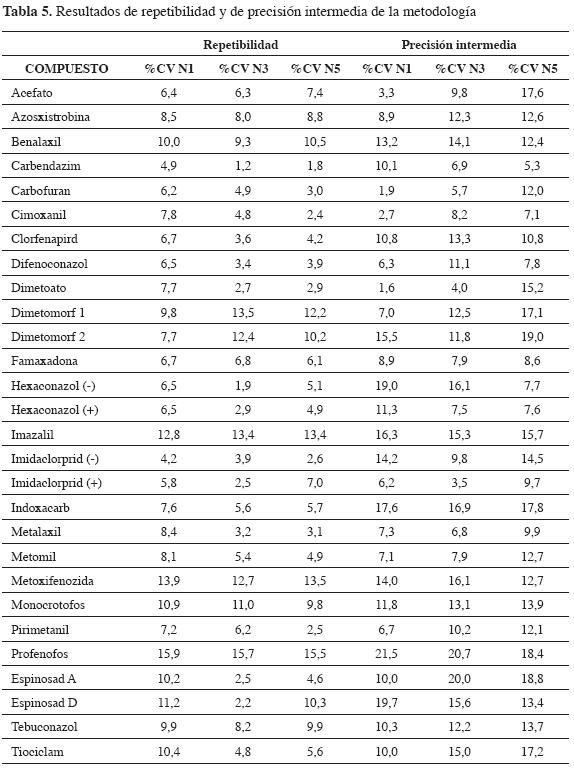
Sin embargo, a pesar de estos aumentos en el tiempo, para todos los compuestos, a excepción de profenofos y espinosad D, se presentan %CV inferiores del 20%, lo cual indica que la metodología es precisa, de acuerdo a las guías de validación de la Unión Europea (17),
Igualmente, se aplicó una prueba de Levene para determinar si existe homogeneidad de varianzas. Los resultados indicaron que, para todos los compuestos, no se presentan diferencias estadísticas entre las varianzas de cada uno de los niveles de concentración en los dos experimentos (repetibilidad y precisión intermedia). De esta manera, se concluye que la precisión de la metodología no varía en función de la concentración (23).
Exactitud
Este parámetro de validación se evaluó determinando el porcentaje de recuperación mediante la fortificación de muestras a tres diferentes concentraciones, alas que se les aplicó el proceso de extracción. Debido a que se presentó homogeneidad de varianzas, es posible promediar los porcentajes de recuperación obtenidos para cada uno de los compuestos en los diferentes niveles de concentración. Las Figura 1 y Figura 2 muestran los porcentajes obtenidos.
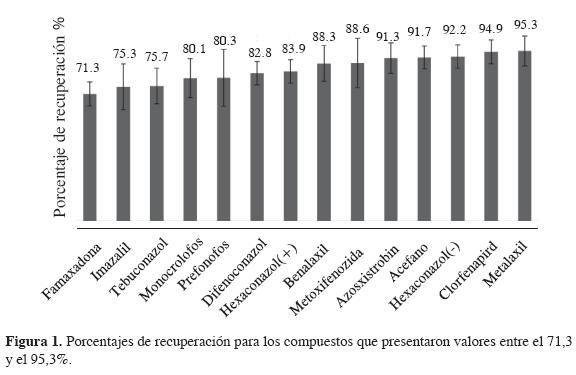
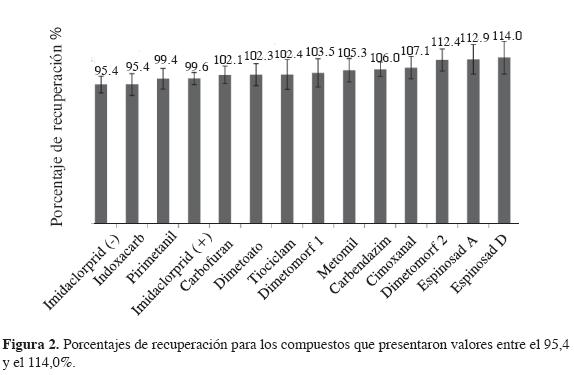
En la Figura 1 se observan los compuestos que presentaron los menores porcentajes de recuperación. Se evidencia que ninguno de los compuestos presenta porcentajes de recuperación inferiores del 70%, límite establecido en la literatura (17). La Figura 2 exhibe los compuestos que presentaron mayores porcentajes de recuperación. Ninguno excede el 120%, con lo cual se concluye que el método es exacto para todos los compuestos (17).
El tiociclam es un compuesto del que diferentes publicaciones y bases de datos especializadas en el análisis de residuos de plaguicidas reportan bajos porcentajes de recuperación; sin embargo, las modificaciones realizadas en este trabajo al método QuEChERS (al asistir la extracción con ultrasonido y emplear acetonitrilo con ácido acético) permiten obtener porcentajes de recuperación superiores (14).
Límites de detección y cuantificación
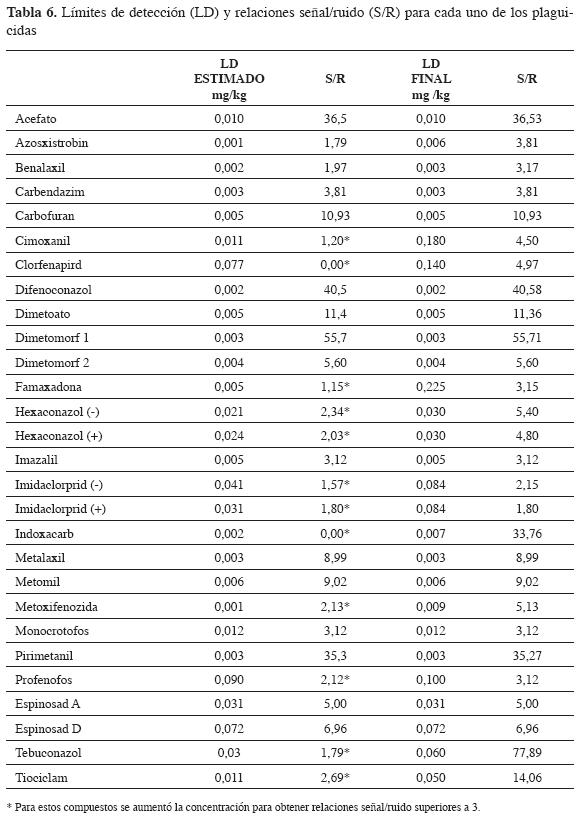
La determinación de los límites de detección y cuantificación se realizó mediante el método propuesto por la IUPAC (15). Se determinó el ruido de varios blancos en las zonas en las que eluyen cada uno de los compuestos, se calculó la desviación estándar y se estimó la concentración de estos límites.
Al tratarse de residuos de contaminantes en alimentos, la determinación de estos límites no solo requiere esta estimación, sino que es necesario comprobar su veracidad. Por consiguiente, para los límites de detección se fortificó un blanco con las concentraciones estimadas y se calculó su relación señal ruido (S/R); en el caso en el que la relación S/R tuvo un valor inferior a tres, se aumentó la concentración hasta obtener relaciones adecuadas. La Tabla 6 muestra los valores límites de detección estimados, la relación señal ruido y los límites de detección propuestos para los compuestos que presentaron señales ruido inferiores a tres.
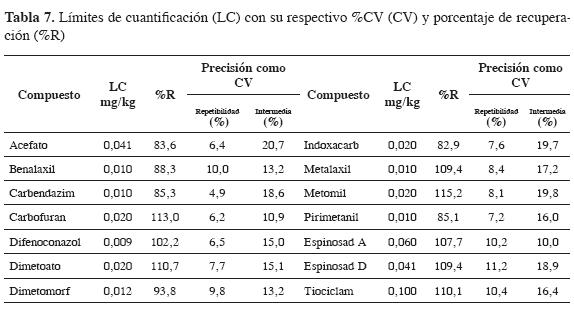
Al realizar la estimación de los límites de cuantificación, muchos de estos coincidían o presentaban valores muy cercanos al punto más bajo de la curva de calibración; por tanto, para estos compuestos se decidió adoptar el valor más bajo de la curva como el límite de cuantificación. Para los compuestos restantes, se fortificaron las muestras al nivel estimado y se calcularon su precisión (como %CV) y su exactitud (como porcentaje de recuperación). La Tabla 7 muestra que para estos compuestos a este nivel de concentración, se encuentra que los porcentajes de recuperación están entre el 70 y el 120%, lo cual indica que a estas concentraciones la metodología es exacta. Sin embargo, el CV del acefato presentó valores superiores al 20%. En los compuestos restantes, la metodología cumple con los criterios establecidos en la literatura (17).
Robustez
La robustez de la metodología se evaluó mediante un diseño de Younden-Steiner y se analizó la influencia de cada factor sobre el porcentaje de recuperación de los plaguicidas. Los parámetros escogidos y sus respectivas variaciones correspondieron a:
1. Masa de sulfato de magnesio: 4 g a 5 g.
2. Masa de PSA: 25 mg por mililitro de extracto a 15 mg por mililitro.
3. Flujo del gas de secado: de 15 a 10 L/min.
4. Flujo del gas nebulizador: 1,5 a 1,0 L/min.
5. Temperatura del bloque de calentamiento: 200 a 300 °C.
6. Temperatura de la línea de desolvatación: 200 a 300 °C.
7. Voltaje del capilar: 4,5 a 3,0 kV y de -4,5 a -3.0 kV.
Este ensayo se realizó en el límite de cuantificación de la metodología. Los resultados mostraron que en la mayoría de los casos, el método es robusto a los cambios en la metodología (primeras tres variables); sin embargo, para los cambios instrumentales realizados (siguientes cuatro variables) se encontró que el método no es robusto para la mayoría de los compuestos.
Las variaciones realizadas a la metodología, en las cuales el método fue robusto, concuerdan satisfactoriamente con los resultados reportados por otros autores (24). Los resultados obtenidos para las variables restantes indican que es de suma importancia su estricto control. La falta de robustez de la metodología a las variables instrumentales se atribuye al complejo proceso de ionización que ocurre en la interfaz, pues estas cuatro variables están íntimamente relacionadas con este proceso. Las variaciones realizadas en estos experimentos fueron bastante significativas, ya que en la mayoría de los casos superaron el 30% del valor original.
Evaluación de la metodología con muestras reales
En la evaluación de las muestras de tomate se encontró un solo compuesto, lo cual se confirmó teniendo en cuenta los parámetros establecidos en el documento guía de la Unión Europea (17). Estos criterios corresponden a la relación de los iones escogidos para la identificación del compuesto (Tabla 1) y a la variación del tiempo de retención del compuesto.
El compuesto identificado mediante la metodología desarrollada correspondió a indoxacarb, el cual presentó una concentración inferior al límite de cuantificación y una relación señal ruido de 15,21 (n = 2).
CONCLUSIONES
Los resultados de la validación realizada mostraron que la exactitud de la metodología multirresiduo desarrollada para el análisis de 24 plaguicidas era adecuada, expresada como porcentaje de recuperación entre el 71,3 y el 114%. En la evaluación de la precisión como repetibilidad, ninguno de los compuestos presentó coeficientes de variación superiores al 15%. Los resultados obtenidos en los experimentos de precisión intermedia indicaron que todos los coeficientes de variación son inferiores al 20%, a excepción de espinosad D y profenofos.
En la evaluación del efecto matriz se evidenció la presencia de este en todos los compuestos estudiados, a excepción de imidaclorprid en modo negativo; en la evaluación de la linealidad se determinó que fue adecuada para todos los compuestos en los rangos de concentración evaluados. El experimento de robustez reveló que el método no es sólido frente a las variaciones instrumentales; por ejemplo, flujo del gas nebulizador, voltaje del capilar y temperaturas de la línea de desolvatación y del gas de secado.
En la evaluación de las muestras provenientes del municipio de Villa de Leyva en el departamento de Boyacá, se encontró indoxacarb en solo una de las 16 muestras analizadas en el periodo evaluado. La cantidad de indoxacarb presente en la muestra es inferior al límite de cuantificación de la metodología y, a su vez, es inferior al límite máximo de residuos.
AGRADECIMIENTOS
Expresamos nuestro agradecimiento al Centro de Biosistemas de la Universidad Jorge Tadeo Lozano de Bogotá y a la Universidad Católica de Lovaina (Bélgica) por su apoyo mediante el proyecto Multidisciplinary assessment of efficiency and sustainability of smallholder-based tomato production systems in Colombia, with a roadmap for change (Código: ZEIN2009PR364).
REFERENCIAS BIBLIOGRÁFICAS
1. Tomlin, C.D. The pesticide manual: A world compendium. Reino Unido, Tomlin C.D. 2009. p. 192. [ Links ]
2. Alba, A. Chromatographic-mass spectrometric food analysis for trace determination of pesticide residues. España, Comprehensive Analytical Chemistry, Elsevier. 2007. p. 215. [ Links ]
3. Snyder, L.R.; Kirkland, J.J.; Dolan, J. Introduction to modern liquid chromatography. Estados Unidos, Wiley. 2009. p. 21. [ Links ]
4. Wang, J.; Leung, D. Applications of ultra-performance liquid chromatography electrospray ionization quadrupole time-of-flight mass spectrometry on analysis of 138 pesticides in fruit- and vegetable-based infant foods. Journal of Agricultural and Food Chemistry. 2009. 57(6): 2162-2173. [ Links ]
5. Pizzutti, I. R.; de Kok, A.; Hiemstra, M.; Wickert, C.; Prestes, O. D. Method validation and comparison of acetonitrile and acetone extraction for the analysis of 169 pesticides in soya grain by liquid chromatography-tandem mass spectrometry. Journal of Chromatography A. 2009. 1216 (21): 4539-4552. [ Links ]
6. Sato, F.; Iwata, H.; Nomura, T.; Komatsu, K. Multiresidue analysis of pesticides in foods using liquid chromatography/mass spectrometry. Estados Unidos, American Chemical Society. 2005. p. 28-37. [ Links ]
7. Soler, C.; Picó, Y. Recent trends in liquid chromatography-tandem mass spectrometry to determine pesticides and their metabolites in food. TrAC Trends in Analytical Chemistry. 2007. 26(2): 103-115. [ Links ]
8. Díaz, L., Llorca-Pórcel, J.; Valor, I. Ultra trace determination of 31 pesticides in water samples by direct injection-rapid resolution liquid chromatography-electrospray tandem mass spectrometry. Analytica Chimica Acta. 2008. 624(1): 90-96. [ Links ]
9. Niessen, W.M.A. Progress in liquid chromatography-mass spectrometry instrumentation and its impact on high-throughput screening. Journal of Chromatography A. 2003. 1000(1-2): 413-436. [ Links ]
10. Anastassiades, M.; Lehotay, S. J.; tajnbaher, D.; Schenck, F. J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and dispersive solid-phase extraction for the determination of pesticide residues in produce. Journal Of AOAC International. 2003. 86(2): 412-431. [ Links ]
11. Furlani, R.P.Z.; Marcilio, K.M.; Tfouni, S.A.V. Determination of pesticides in sugarcane juice using the quick, easy, cheap, effective, rugged and safe (QuEChERS) sample preparation. Toxicology Letters. 2008. 180(Supplement 1): S75-S75. [ Links ]
12. Koesukwiwat, U.; Sanguankaew, K.; Leepipatpiboon, N. Rapid determination of phenoxy acid residues in rice by modified QuEChERS extraction and liquid chromatography-tandem mass spectrometry. Analytica Chimica Acta. 2008. 626(1): 10-20. [ Links ]
13. Lesueur, C.; Knittl, P.; Gartner, M.; Mentler, A.; Fuerhacker, M. Analysis of 140 pesticides from conventional farming foodstuff samples after extraction with the modified QuECheRS method. Food Control. 2008. 19(9): 906-914. [ Links ]
14. Ahumada, D.A.; Zamudio, A. M. Development of a method for determination of thiocyclam and other pesticides by UFLC/MS. 3rd Latin American Pesticide Residue Workshop Food and Environment. Uruguay, Universidad de la República. 2011. p. 99. [ Links ]
15. Lee, P.W. Handbook of residue analytical methods for agrochemicals. Reino Unido, John Wiley & Sons. 2003. p. 352. [ Links ]
16. Dejaegher, B.; Heyden, Y.V. Ruggedness and robustness testing. Journal of Chromatography A. 2007. 1158(1-2): 138-157. [ Links ]
17. SANCO. Method Validation and quality Control Procedures for pesticide residues analysis in food and feed. 2009. [citado: 2011 22 julio]; Disponible en http://ec.europa.eu/food/plant/protection/resources/qualcontrol_en.pdf. [ Links ]
18. Ahumada, D.A.; Guerrero, J.A. Study of matrix effect in pesticide analysis by gas chromatography. VITAE. 2010. 17(1): 51-58. [ Links ]
19. Kruve, A.; Künnapas, A.; Herodes, K.; Leito, I. Matrix effects in pesticide multi-residue analysis by liquid chromatography-mass spectrometry. Journal of Chromatography A. 2008. 1187(1-2): 58-66. [ Links ]
20. España, J.C.; Zamudio, A.M.; Ahumada, D.A. Studies on matrix effects in liquid chromatography-mass spectrometry, 3rd Latin American Pesticide Residue Workshop Food and Environment. Uruguay, Universidad de la República. 2011. p. 227. [ Links ]
21. Hadi, S.C. Regression Analysis by Example. 4th ed. New Jersey, John Wiley & Sons. 2006. p. 129. [ Links ]
22. Kleinbaum, L.L.K. Applied regression analysis and multivariable methods. 2nd ed. Pacific Grove, US, Brooks/Cole Publishers.1989. p. 227. [ Links ]
23. Soboleva, N.R.; Mageto, Á.; Ambrus, A.; Fajgelj, A. Principles and practices of method validation. Cambridge, UK, Royal Society Of Chemistry. 2000. p. 171. [ Links ]
24. Wong, J.; Hao, C.; Zhang, K.; Yang, P.; Banerjee, K.; Hayward, D.; Iftakhar, I.; Schreiber, A.; Tech, K.; Sack, C.Development and Interlaboratory Validation of a QuEChERS-Based Liquid Chromatography- Tandem Mass Spectrometry Method for Multiresidue Pesticide Analysis. Journal of Agricultural and Food Chemistry. 58(10): 5897-5903. [ Links ]

