










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804
Rev.Colomb.Quim. vol.41 no.3 Bogotá Sept./Dec. 2012
Proton transfer from 1,4-pentadiene to superoxide radical anion: a QTAIM analysis
Transferencia de protón desde el 1,4-pentadieno al radical anión superóxido: un análisis QTAIM
Transferência de prótons de 1,4-pentadieno o ânion radical superóxido: uma análise QTAI
Ángela Rodríguez-Serrano,1 Martha C. Daza,1* Markus Doerr,1,2 José Luis Villaveces3
1 Grupo de Bioquímica Teórica, Escuela de Química, Universidad Industrial de Santander, Carrera 27, Calle 9, Bucaramanga,Colombia
2 Facultad de Química Ambiental, Universidad Santo Tomás, Carrera 18 No. 9 - 27, Bucaramanga, Colombia
3 Grupo de Química Teórica, CEIBA, Universidad de los Andes, Carrera 1 No. 18A - 12, Bogotá, Colombia
* Corresponding author: mcdaza@correo.uis.edu.co
Recibido: 25/08/12 - Aceptado: 27/11/12
Abstract
We studied the bis-allylic proton transfer reaction from 1,4-pentadiene to superoxide radical anion (O2−). Minima and transition state geometries, as well as thermochemical parameters were computed at the B3LYP/6-311+G(3df,2p) level of theory. The electronic wave functions of reactants, intermediates, and products were analyzed within the framework of the Quantum Theory of Atoms in Molecules. The results show the formation of strongly hydrogen bonded complexes between the 1,4-pentadien- 3-yl anion and the hydroperoxyl radical as the reaction products. These product complexes (PCs) are more stable than the isolated reactants and much more stable than the isolated products. This reaction occurs via pre-reactive complexes which are more stable than the PCs and the transition states. This is in agreement with the fact that the net proton transfer reaction that leads to free products is an endothermic and nonspontaneous process.
Key words: superoxide radical anion, density functional theory, QTAIM, reaction mechanism, proton transfer,1,4-pentadiene
Resumen
Nosotros estudiamos la reacción de transferencia de protón bis-alílico del 1,4-pentadieno al radical anión superóxido (O2−). Las geometrías de los mínimos y de los estados de transición, así como también los parámetros termoquímicos se calcularon usando el nivel de teoría B3LYP/6-311+G(3df,2p). Las funciones de onda electrónicas de los reactantes, intermedios y productos se analizaron dentro del marco de la teoría cuántica de átomos en moléculas. Nuestros resultados muestran la formación de complejos estabilizados por enlaces de hidrógeno entre el anión 1,4pentadien-3-ilo y el radical hidroperoxilo como productos de reacción. Estos complejos producto (PCs) son más estables que los reactantes aislados y mucho más estables que los productos aislados. Esta reacción ocurre vía la formación de complejos pre-reactivos, los cuales son más estables que los PCs y los estados de transición. Estos resultados están de acuerdo con el hecho de que la reacción global de transferencia de protón que conduce a la formación de los productos libres es un proceso endotérmico y no espontáneo.
Palabras clave: radical anión superóxido, teoría del funcional de la densidad, QTAIM, mecanismos de reacción, transferencia de protón, 1,4-pentadieno
Resumo
Estudou-se a reação de transferência do próton bis-alílico do 1,4-pentadieno ao radical ânion superóxido (O2−). As geometrias dos mínimos e dos estados de transição, bem como os parâmetros termoquímicos foram calculadas utilizando o nível de teoria B3LYP/6-311+G(3df, 2p). As funções de onda eletrônica dos reagentes, intermediários e produtos foram analisadas no âmbito da teoria quântica de átomos em moléculas. Os resultados obtidos demonstram a formação de complexos estabilizados por ligações de hidrogênio entre o ânion 1,4-pentadieno- 3-ilo e o radical hidroperoxilo como produtos de reação. Estes complexos formados como produtos (PCs) são mais estáveis do que os reagentes isolados e muito mais estáveis do que os produtos isolados. Esta reação ocorre por meio de complexos pré-reativos mais estáveis do que os PCs e os estados de transição. Estes resultados estão de acordo com o fato da reação global de transferência de próton que conduz à formação dos produtos livres, é um processo endotérmico e não espontâneo.
Palavras-chave: radical ânion superóxido, teoria do funcional da densidade, QTAIM, mecanismos de reação, transferência de próton, 1,4-pentadieno
INTRODUCTION
The superoxide radical anion is the product of one-electron reduction of molecular oxygen and plays important roles in atmospheric chemistry (1-2), photochemical (3) and in many biochemical processes (4-5). The generation of the superoxide radical anion is known to be a key process in the decomposition of ozone in cloud water droplets and is involved in the regulation of the overall oxidation processes of the troposphere (6). Moreover, in living cells the superoxide radical anion is a byproduct of the mitochondrial respiratory chain, where it is usually generated in processes involving NADPH oxidases,(7) mitochondrial electron transport, (8) and cytochrome P-450 reactions(9). It is thought to be important in the radical theory of aging (10).
The established chemistry of the superoxide radical anion does not define it as a strong biological oxidant. Nevertheless, it can be considered as a source of other reactive oxygen species (ROS i.e. H2O2, HO•, HO−, HOO•, 1O2) which can potentially cause cell damage. Disproportionation of superoxide radical anion by superoxide dismutase (SOD) produces hydrogen peroxide and molecular oxygen. Highly reactive hydroxyl radicals can be produced by the decomposition of hydrogen peroxide via Fenton and Haber-Weiss reactions. The protonation of the superoxide radical anion leads to the formation of its conjugated acid, the hydroperoxyl radical, equation [1] (11).
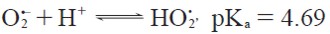 | [1] |
Although the superoxide radical anion is not as reactive as the other ROS, it has a dual nature, i.e., it is a negatively charged radical and it bears an unpaired electron. Thus, for understanding its reactivity it is important to consider its capability of inducing free radical reactions, its basicity and nucleophilicity, and how well it acts as either a oneelectron reducing agent or an oxidant. Indeed, the superoxide radical anion is relatively unreactive in typical free radical reactions such as hydrogen atom abstraction or addition to double bonds (12). Because the effective basicity of the superoxide radical anion is quite high, it preferably acts as a Brønsted base rather than as a hydrogen atom acceptor in reactions with labile hydrogens. The C-H bonds with low pKa values undoubtedly react with superoxide radical anion via initial proton transfer. Thus, Stanley et al (13) report that cyclopentadiene (pKa = 16) and diethyl malonate (pKa = 13) induce instantaneous disproportionation of superoxide radical anion. Dibenzoylmethane (pKa = 9.4 (14)) and 1,3-cyclohexadione (pKa = 4.8 (15)) are also rapidly deprotonated by superoxide radical anion, though the resulting anions are stable to oxygen and superoxide radical anion (16). Similarly, the oxidations of benzoylacetonitrile (17) (pKa = 7.8) malonitrile (pKa = 11.2), benzyl cyanide (18) (pKa = 22) and carbonylic compounds (17) (pKa ≈ 20) at the methylene α-position are expected to be initiated by proton transfer reactions, leading to the respective anion plus the hydroperoxyl radical, equation [2]:
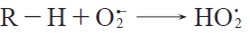 | [2] |
While hydroperoxyl radical can selectively abstract bis-allylic hydrogen from 1,4-polyunsaturated fatty acids, the superoxide radical anion cannot (11). Due to the lack of charge the hydroperoxyl radical is more soluble in key sites of oxidant reactivity such as the hydrophobic core of biological membranes. On the other hand, since the redox potential of O2/O2− and O2.−/H2O2 are −0.7 and 1.3 V at pH = 7, the superoxide radical anion frequently acts as a reductant in the presence of electron acceptors and as an oxidant in the presence of electron donors. In other words, its reactivity depends on the chemical nature of the substrate. Thus, further studies of the interactions and reactivity of the superoxide radical anion with different kinds of substrates are expected to improve the understanding of the mechanisms as well as, in general, which kind of substrates would promote either redox, radical or proton transfer reactions.
Theoretical investigations have been proven useful to improve the understanding of reaction mechanisms. Moreover, theoretical methods are sometimes the only tool for studying unstable com pounds and short-lived intermediates generated in many chemical reactions, as well as reaction mechanisms. Surprisingly, in contrast to the wealth of earlier experimental studies that are available for superoxide radical anion, only few theoretical studies have been made on the chemical reactivity of this specie, mainly hydrogen atom and proton transfer processes. For example, it has been found that the superoxide radical anion can induce a proton transfer from H2S to form a very stable HS-HO2 complex which has been found both experimentally and theoretically (19). Moreover, hydrogen atom abstraction from quercetin by superoxide radical anion has been suggested to follow a coupled hydrogen-electron transfer: first a hydrogen atom is transferred from quercetin to the superoxide radical anion, and then the electron is transferred from the superoxide radical anion to the quercetin, resulting in a net proton transfer reaction (20).
The formation of a stable hydrogen bond complex between hydroperoxyl radical and methanetiol anion (stabilization energy ∼40 kJ/mol) has been proposed as the first intermediate step of the complete oxidation of methanetiol by the superoxide radical anion, the total proton transfer being a non-spontaneous reaction. A similar behavior has been found in studies on the cysteine oxidation by superoxide radical anion (21).
Furthermore, Villamena et al (23-24)have used theoretical tools and stoppedflow kinetics for the rational design of EPR spin traps for superoxide radical anion by studying its nucleophilic characteristics upon addition to the nitronyl carbon atom of nitrones, a reaction which is very important for the understanding of free-radical mediated processes (25-29).
In our previous work we have made a detailed analysis of the C−O hydrogen bonds between 1,4-pentadiene and superoxide radical anion (30). In the present study, we shed more light on the interactions of the superoxide radical anion with 1,4-pentadiene in a proton transfer reaction. 1,4-pentadiene was chosen as a model of the bis-allylic region of a polyunsaturated fatty acid. Though the net bis-allylic proton transfer from 1,4-pentadiene (pKa = 30 (31-32)) to the superoxide radical anion is an unfavorable process, our results show the formation of a very stable hydrogen bonded complex between the 1,4-pentadien-3-yl anion and the hydroperoxyl radical, which is the product of the reaction. Moreover, the distribution of the electronic density associated with the formation and breaking of bonds, as well as the ionic or covalent nature of the atomic interactions have been analyzed within the framework of the Quantum Theory of Atoms in Molecules (QTAIM).
COMPUTATIONAL METHODS
Geometry optimization and thermochemical parameters
Computations of electronic energies, optimizations of the molecular geometries and calculations of vibrational frequencies were carried out using unrestricted density functional theory (UDFT), employing the B3LYP (33-34) functional with the 6-31+G(2d,p) and the 6-311+G(3df,2p) basis sets as implemented in Gaussian 03 (35). Minima and transition states were characterized by having zero and one imaginary vibrational frequencies, respectively. Fully optimized transition state structures were confirmed to be connected to the desired pre-reactive and product complexes by performing an intrinsic reaction coordinate (IRC) scan at the B3LYP/6-31+G(2d,p) level of theory.
The global standard reaction enthalpies (ΔH°) and Gibbs's standard free energies (ΔG°) were obtained from the calculated thermal, entropic and zeropoint energy corrections using unscaled frequencies as shown in equation [3] - [4]:
 | [3] |
 | [4] |
where ε0 is the total electronic energy without the zero-point energy correction and Hcorr and Gcorr are defined as follows:
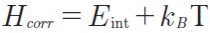 | [5] |
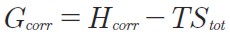 | [6] |
Eint is the correction to the internal thermal energy (including vibrational, translational and rotational contributions), kB is the Boltzmann´s constant, T is temperature (298 K) and Stot is the sum of total entropic contributions from vibrational, rotational and t ranslational motion, respectively. Moreover, the bond dissociation energies (BDE) of the 1,4-pentadiene bis-allylic C-H bonds and the electron, proton and hydrogen affinities (EA, PA, HA) of the superoxide radical anion were also analyzed. The BDEs of the C-H bis-allylic 1,4-pentadiene were computed setting the electronic energy of hydrogen atom to its exact value of -0.5 hartree.
Relative energies (ΔE0), standard activation free energies (Δ‡G°), and activation enthalpies (Δ‡H°) of pre-reactive complexes, transition states and product complexes were computed relative to the energies of the reactants (1,4-pentadiene and superoxide radical anion) at infinite separation. In addition the activation energies were also calculated relative to the pre-reactive complexes for comparison. The binding energies of the pre-reactive and product complexes were calculated according to equations [8] - [9]:
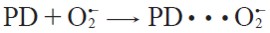 | [8] |
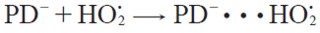 | [9] |
where PD denotes1,4-pentadiene, PD- the 1,4-pentadien-3-yl anion and PC the product complex. Furthermore, the binding energies of the pre-reactive complexes were corrected for the basis set superposition error (BSSE). The counterpoise corrected energies (ΔECP) were obtained using the procedure of Boys and Bernardi (36). To calculate ΔECP and ΔE, the supermolecule (AB) was considered to be made up of two interacting systems A (1,4-pentadiene) and B (superoxide radical anion) as shown in equation [10]:
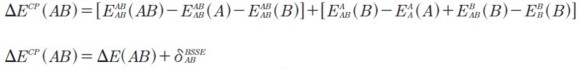 | [10] |
where EXY (Z)represents the energy of Z at the geometry Y calculated with the basis X, ΔECP(AB) denotes the terms in the first parentheses in equation [10] and δBSSEAB is the counterpoise correction (the terms in the second parentheses in equation [10]).
TOPOLOGICAL ANALYSIS OF THE ELECTRON DENSITY
The proton transfer reaction between 1,4-pentadiene and superoxide radical anion was analyzed within the framework of the Quantum Theory of Atoms in Molecules (QTAIM) (37), locating and characterizing the electron density (ρ(r)) at the bond critical points (BCPs). The local properties at the bond critical points such as ρ(r), ∇2ρ(r) (the Laplacian of the electron density), its shape measured by the ellipticity ε, the dimensionless ratio |V(r)|/G(r) of the densities of the potential and the kinetic energies (see below), and the total energy density H(r) were used in order to describe the rupture and formation of bonds. ∇2ρ(r) indicates whether electron density is locally concentrated or depleted. In general, according to the sign of ∇2ρ(r) at the BCP, atomic interactions can be classified as closed shell (CS, ∇2ρ(r) > 0) or shared shell (SS, ∇2ρ(r) < 0).Closed shell interactions are typically found in interactions between ions, such as in NaF, but also in hydrogen bonds. Shared shell interactions, the other extreme, are found between covalently bound atoms, such as the two C atoms in ethane (38-40).
More information about the relative strengths of the interatomic interactions can be obtained by means of the total energy density at the BCP, H(r) = G(r) + V((r), where G(r) and V(r) are the kinetic and the potential energy densities at the corresponding BCP. Because always G(r) > 0 and V(r) < 0, the interactions for which ∇2ρ(r) < 0 are dominated by a local reduction of the potential energy and those interactions for which ∇2ρ(r) > 0 are accompanied by a local excess in kinetic energy. Negative values of H(r) are associated with stabilizing charge concentration in the internuclear region. Its magnitude reflects the "covalence" of a given interaction. Taking into account the values of H(r) at the BCP, the atomic interactions are divided into three regions: pure closed shell (region I, ∇2ρ(r)> 0 and H(r) > 0), transit closed shell(region II, ∇2ρ(r) > 0 and H(r) < 0) and pure shared shell (region III, ∇2ρ(r) < 0 and H(r) < 0). The boundaries of these regions are defined by H(r) = 0 (i.e., |V(r)|/G(r) = 1) and |V(r)|/G(r) = 2 Inside region I, the bond degree BD = |V(r)|/G(r) is an index of non-covalent interactions, quantifying a softening degree (SD) per electron density unit at the BCP. The weaker the interaction the greater is the SD magnitude. In regions II and III, BD measures the degree of covalence (CD) of any pairwise interaction: the stronger the interaction, the greater the magnitude of CD (41).
Furthermore, the change of the atomic properties such as the total energy E(Ω), the potential energy V(Ω), the volume v(Ω), the net charge q(Ω) and the dipolar polarization |M(Ω)| integrated over the atomic basins Ω of the H, C, and O atoms directly involved in theproton transfer reaction were analyzed. According to Popelier´s criteria, after the formation of a hydrogen bond there should be a loss of q(Ω), an energetic destabilization, a decrease of |M(Ω)|and a decrease of v(Ω) of the H atom (30-31).These five criteria were also applied to analyze the atomic interactions between the superoxide radical anion and 1,4-pentadiene at the reaction site and the atomic contributions to the stabilization of each reaction intermediate.
The topological analyses of ρ(r) were carried out using the AIM2000 program(42) with all options set to their default values and the wave functions obtained at the UB3LYP/6-311+G(3df,2p) theoretical level.
NATURAL POPULATION
ANALYSIS
Spin densities (ρ) and charge densities (q) were obtained from a Natural Population Analysis (NPA) (43-44) at the UB3LYP/6-311+G(3df,2p) optimized geometries. The NPA was performed using the program NBO 5.0 (45).Partial charges and partial spin densities were computed for all reaction intermediates in the gas phase. The analysis was made by calculating the sum of charges and spin densities of each atom of the hydrocarbon and oxygen fragment.For the pre-reactive complexes, the hydrocarbon fragment corresponds to 1,4-pentadiene and the oxygen fragment to superoxide radical anion. For product complexes, the oxygen fragment is the hydroperoxyl radical and the hydrocarbon fragment is the 1,4-pentadien-3-yl anion.
RESULTS AND DISCUSSION
Potential energy surfaces of 1,4-pentadiene, penta-1,4-dien-3-yl anion and penta-1,4-dien-3-yl radical
First, we characterized the potential energy surface of 1,4-pentadiene taking into account symmetry. According to our previous work, three minima are found (30). The global minimum has C2 symmetry and the two local minima are of C1- envelope and CS symmetry, see Figure 1. This PES is relatively flat, with an energy difference between the highest and lowest points of only 11.7 kJ/mol. This means that the rotation around the two single bonds of 1,4-pentadiene is barrierless(30).

The gas phase bis-allylic proton transfer from 1,4-pentadiene to superoxide radical anion leads to the penta-1,4- dien-3-yl anion. The PES of the 1,4-pentadien- 3-yl anion PES has three minima: a global minimum with CS symmetry and two local minima with C1-envelope and C2 symmetry, Figure 2. The energy differences between the global minimum and the C1-envelope and C2 conformers are 10.7 and 11.0 kJ/mol at the UB3LYP/6- 311+G(3df,2p) level of theory.
A 1,4-pentadien-3-yl radical is produced when a bis-allylic hydrogen atom abstraction from 1,4-pentadiene occurs. We found three minima on the global minimum and two local minima with C1-envelope and C2 symmetry, Figure1. The findings are in agreement with the theoretical work of Szoriand Viskolcz (46).The energy differences between the global minima and the C1-envelope and C2 conformers are 6.0 and 21.1 kJ/mol computed with the 6-311+G(3df,2p) basis set. Moreover, it has been demonstrated by EPR spectroscopy that two planar conformations of 1,4-pentadien-3-yl radicals,C1-envelope and Cs, are in equilibrium at room temperature and above (the difference of the experimental enthalpy of formation of these conformations is about 4.2 kJ/mol (47)). The third possible planar isomer, the C2 conformer, is not significantly populated at room temperature (48).
GLOBAL REACTION PARAMETERS
The computed C-H bis-allylic bond dissociation energies (BDEs), which characterize the hydrogen and proton donating ability of 1,4-pentadiene leading to the formation of 1,4-pentadien-3-yl radical, are depicted in Table 1. Minor deviations from the experimental value(320.5 kJ/mol) (49) were found for th C-H bis-allylic bond dissociation energies calculated for the three minima of 1,4-pentadiene. These values are lower than the BDE for the homolysis of a methyl C-H bond of propene (369 kJ/mol) (50) and higher than the BDE for the cyclopentadiene methylene C-H bond(297.5 kJ/mol) (51). On the other hand,the formation of the 1,4-pentadien-3-yl anion is more endergonic than the radical formation, indicated by its higher proton affinity (1366.8 - 1355.6 kJ/mol). This is expected taking into account the high pKa value (pKa≈ 30)(31-32) of the bisallylic proton and the resonance stabilization of the anion.
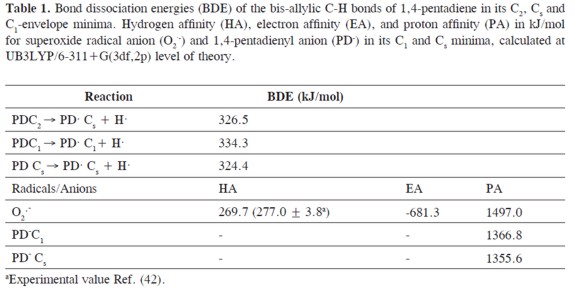
The overall standard reaction enthalpies(ΔH°), the Gibbs´s standard free energies (ΔG°) and the reaction energies(ΔE0) for the proton transfer reaction calculated in the gas phase are listed in Table 2. According to these results, the gas phase proton transfer from 1,4-pentadiene to superoxide radical anion is endergonic, endothermic, and non-spontaneous. Comparing these reactions to a hypothetical hydrogen abstraction reaction from 1,4-pentadiene by superoxide radical anion, these parameters are similar for both kinds of reactions in the gas phase. Because the calculated gas phase proton affinity of the superoxide radical anion of 1497.0 kJ/mol is higher than itshydrogen affinity of 269.7 kJ/mol (Table 1), the proton transfer reaction is preferred over the hydrogen abstraction. This is also supported by its negative electron affinity of -681.3 kJ/mol. The calculated hydrogen affinity of superoxide radical anion (at the UB3LYP/6-311+G(3df,2p)theoretical level) is in excellent agreement with the experimental value (277.0 ± 3.8 kJ/mol) (52).

It is known that C-H bonds with low pKa values like cyclopentadiene (pKa =16) and diethyl malonate (pKa= 13) induce instantaneous disproportionation of superoxide radical anion (13). Because the pKa of 1,4-pentadiene is almost two times higher (pKa≈30) (31-32) than the pKa of cyclopentadiene and diethyl malonate (13) its proton transfer to superoxide radical anion is non-spontaneous.
This is consistent with the standard Gibbs energies calculated for this reaction in the gas phase of 37.9 and 46.7 kJ/mol.
MECHANISM OF TRANSFER FROM 1,4-PENTADIENE TO SUPERXIDE RADICAL ANION
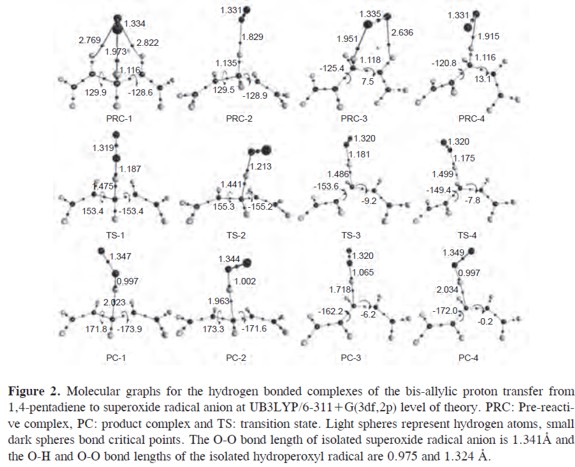
Four reaction pathways for the bisallylic proton transfer from 1,4-pentadiene to superoxide radical anion were found. The molecular graphs and some selected geometrical parameters of the minima and transition states obtained at the UB3LYP/6-311+G(3df,2p) theoretical level are shown in Figure 2, and the energy reaction profiles in Figure 3. The pre-reactive complexes, transition states and product complexes are designated by the prefixes PRC, TS and PC, respectively, followed by a number to differentiate different pathways. The free reactants and free products are labeled with the prefixes FR and FP.
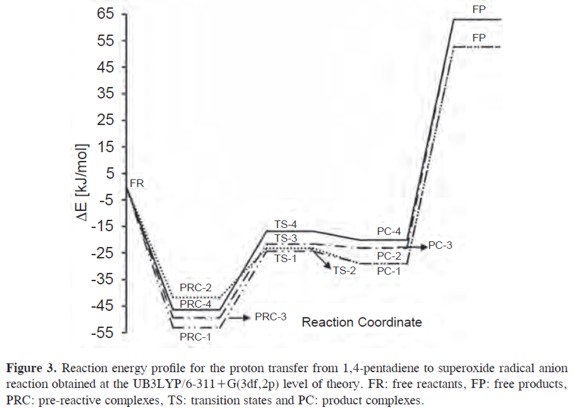
The proton transfer reaction from 1,4-pentadiene to superoxide radical anion starts with the formation of the pre-reactive complexes between superoxide radical anion and 1,4-pentadiene prior to the transition states, see equation [11 ] - [12]. We assume that the pre-reactive complexes are in equilibrium with the free reactants.
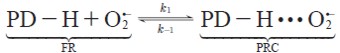 | [11] |
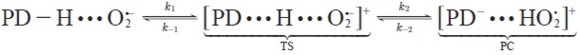 | [12] |
In our previous work, twelve hydrogen bonded complexes have been found on the PES of the superoxide radical anion and 1,4-pentadiene at the UB3LYP/6-311+G(3df,2p) level of theory (30). Three of them were found to be pre-reactive complexes (PRC-1, PRC-3 and PRC-4) for the proton transfer reaction. The counterpoise corrected binding energies of PRC-1, PRC- 2, PRC-3 and PRC-4 are -48.9, -38.0,-45.3 and -42.3 kJ/mol (see Table 3),respectively with a BSSE correction of less than 8%. The 1,4-pentadiene in the PRC-1 and PRC-2 complexes has nearly Cs symmetry, and in PRC-3 and PRCPD 4 it nearly has C1-envelope symmetry. A proton transfer pathway from the C2 symmetric minimum of 1,4-pentadiene was not found.
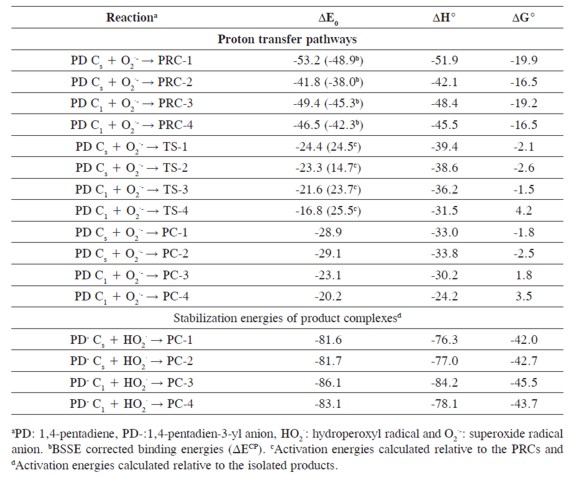
The proton transfer reaction paths through the PRC-1, PRC-2, PRC-3 and PRC-4 pre-reactive complexes are connected to the transition states TS-1, TS-2, TS-3 and TS-4, respectively. In Table 3, the activation energies (ΔE0), standard enthalpies (ΔH°) and the Gibbs's standard free activation energies (ΔG°) for these transition states are shown. All transition states have negative activation energies, activation enthalpies and low Gibbs´s activation free energies. Nevertheless, the formation of these transition states from the pre-reactive complexes requires 24.4, 14.7, 23.7 and 25.5 kJ/mol, respectively, see Table 3. These transition states are all more stable than the isolated reactants and energetically less stable than the pre-reactive complexes. The imaginary frequencies which characterize each transition state are also listed in Figure 2. These imaginary frequencies correspond almost exclusively to the bis-allylic C-H bond breaking.
As the Hammond postulate states, when a transition state leading to an unstable reaction intermediate (or product) has nearly the same energy as that intermediate, the two intermediates could be interconverted with only a small reorganization of their molecular structure (53). This means that the transition state bears the larger geometrical resemblance to the less stable species (product or reaction intermediate). Taking into account the endothermic global reaction enthalpies for the proton transfer to superoxide radical anion and the negative activation energies, the transition state resembles the products and is called a "late" transition state. This is reflected in the significant increase of the C-H bond length(34-35%) and the shortening of the O-H bond (21-22%), i.e., the transition states are late due to their long C-H and short O-H bond lengths.
It follows that strongly hydrogen bonded complexes between the products, the 1.4-pentadien-3-yl anion and the hydroperoxyl radical, have to be considered as the final products of the bis-allylic proton transfer from 1,4-pentadiene to superoxide radical anion, see equation [12]. These product complexes PC-1, PC-2, PC-3 and PC-4 have a relative energy of -24.4, -23.3, -21.6 and -16.8 kJ/mol, considering the energy difference between them and the isolated reactants (1,4-pentadiene and superoxide radical anion). In all cases the product complexes are less stable than the prereactive complexes by 23.3, 11.5, 25.3 and 25.0 kJ/mol and the energy difference between the product complexes and the transition states is very small (4.5,5.8, 1.5 and 3.4 kJ/mol). This means that the reverse reaction to the pre-reactive complexes would be expected to compete with the direct reaction. Nevertheless,the strong binding energies of the product complexes (-81.6 and -86.1 kJ/mol, equation [9]) are considerably higher than the binding energies of the pre-reactive complexes (-53.2 and -49.4kJ/mol), Table 1. According to these results, once the product complexes are formed, the backward reaction that may lead to the pre-reactive complexes and the release of the free products would not be possible, equation [13].
 | [13] |
NPA CHARGES AND SPIN DENSITIES
In Table 4 we show the charges (q)and spin densities (ρ) calculated at the hydrocarbon and oxygen containing fragments for the pre-reactive complexes, transition states and product complexes. The charges and spin densities calculated for each fragment of the pre-reactive complexes confirm that those complexes are composed of a superoxide radical anion (q ≈ -0.9 and ρ ≈ 1) and 4-pentadiene(q ≈ -0.1 and ρ ≈ 0). Furthermore, the hydrocarbon and oxygen fragments at the transition states TS-1, TS-2, TS-3 and TS-4 are both negatively charged and the spin densities are almost zero for the hydrocarbon fragment and ≈ 1 for the oxygen fragment. The transferred proton is nearly half positively charged at the transition states (≈ 0.4 a.u.) and has a spin density of almost zero.
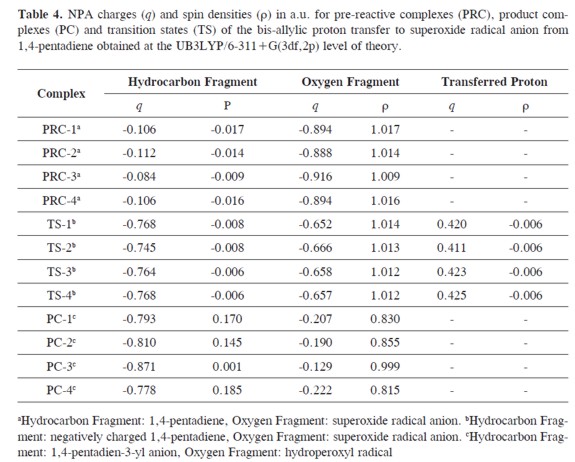
Finally, spin densities calculated at the oxygen fragment of the product complexe PC-1, PC-2, PC-3 and PC-4 are almost 1 a.u. and the charges at the hydrocarbon fragment are in the range of -0.87 and -0.79 a.u. These results confirm the proton transfer character of the reaction between the superoxide radical anion and the 1,4-pentadiene and that these product complexes are composed of the 1,4-pentadien-3-yl anion and the hydroperoxyl radical.
TOPOLOGICAL ANALYSIS OF THE ELECTRON DENSITY OF THE HYDROGEN BONDED COMPLEXES
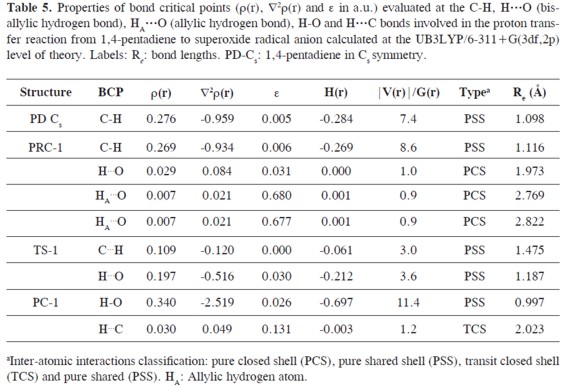
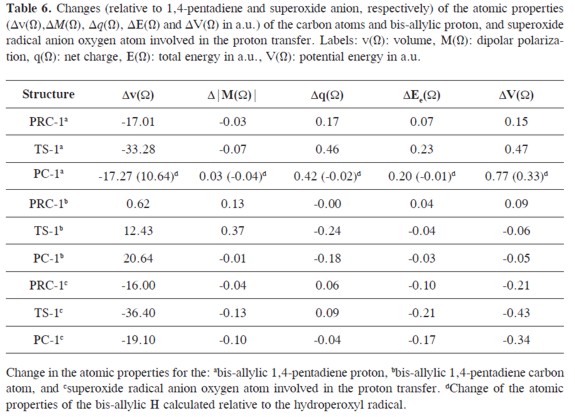
The electron density ρ(r), the Laplacian of the electron density ∇2ρ(r), the ellipticity ε, total energy density H(r)and the |V(r)|/G(r) ratio calculated at the bond critical points (3,-1) of the 1,4-pentadiene-superoxide radical anion hydrogen bonded complexes and their equilibrium distances (Re(Å)) are shown in Table 5. Furthermore, the changes of the properties integrated over the atomic basins (Ω) of the atoms involved in the proton transfer reaction are also reported in Table 6. In the course of the reaction, the changes of the atomic properties are analyzed taking into account the difference of the atomic property in the complex (PRC, TS or PC) relative to the values of isolated 1,4-pentadiene and superoxide radical anion. In addition, the molecular graphs of the hydrogen bonded complexes involved in the proton transfer reaction are presented in Figure 2. The atomic properties which are considered here are the changes in the atomic total integrated volume (Δv(Ω), 0.001 a.u. isosurfaces), the first moments of the atomic charge distribution (Δ|M(Ω)|), the atomic charges (Δq(Ω)), and the atomic kinetic (ΔEe(Ω)) and total potential energies (ΔV(Ω)).
For the sake of simplicity, we only discuss the topological analysis of the electron density at the C-H, H···O (bisallylic hydrogen bond), HA···O (allylic hydrogen bond), H-O and H···C bond critical points at the PRC-1, TS-1 and PC-1 structures, because the tendencies for the other pathways are the same.
LOCAL TOPOLOGICAL PROPERTIES
The superoxide radical anion can make hydrogen bonds with the allylic (HA) and bis-allylic (H) hydrogen atoms of 1,4-pentadiene. In general the C-HA···O allylic hydrogen bonds are weaker than the C-H···O bis-allylic hydrogen bonds (30). PRC-1 is a cyclic complex stabilized by both, allylic and bis-allylic hydrogen bonds. The bis-allylic H···O hydrogen bond length (1.973 Å) is shorter than the allylic HA···O hydrogen bonds (2.769- 2.822 Å). The HA···O ellipticities (ε) at the BCP are ≈ 0.68 a.u. while the bisallylic bond ellipticities are close to zero (0.031 and 0.048 a.u., respectively).Since it has been shown that ρ(r) is related to the bond strength, it is clear that the C-HA···O hydrogen bonds in these complexes are weaker (ρ(r) between 0.007 and 0.008 a.u.) than the C-H···O bis-allylic hydrogen bonds (ρ(r) of 0.029 and 0.030 a.u.). Furthermore, the strengthening of the bis-allylic H···O hydrogen bond driven by the shortening of the hydrogen bond length and the elongation of the bis-allylic 1,4-pentadiene C-H bond leads to the formation of TS-1 and PC-1.
In general, it can be seen that ρ(r) at the C-H bond diminishes when going from PRC-1 to PC-1 (0.269, 0.109 and 0.030 a.u.). The sign of ∇2ρ(r) changes from negative values at PRC-1 and TS-1 (-0.934, -0.120 a.u.) to positive values at PC-1 (0.049 a.u.). The low value of ρ(r)(0.030 a.u.) and the positive Laplacian ∇2ρ(r) (0.049 a.u.) at the C···H BCP of PC-1 is an indication of a depletion of the charge density along the bond path, which is characteristic of a closed shell interaction and an evidence of the rupture of the covalent 1,4-pentadiene C-H bond. On the other hand, the bis-allylic O···H BCP undergoes a transition from a hydrogen bond in the PRC-1 to a covalent O-H bond in the PC-1. This situation is reflected by the change of the sign of ∇2ρ(r) being positive at PRC-1 (0.084 a.u.) and negative at the TS-1 and PC-1(-0.516 and -2.519 a.u). Moreover, an increase of ρ(r) at the O···H BCP from 0.029 a.u. at the PRC-1 to 0.340 a.u. at the PC-1 is observed.
The shape of the electron density distribution in a plane through a BCP and perpendicular to the bond is measured by its ellipticity ε. It is defined in terms of the negative eigenvalues (or curvatures) of the Hessian of ρ(r), λ1 and λ2, as ε = (λ1/λ2) = 1 and also is a measure of the extent to which electron density is accumulated in a given plane. The ellipticities at the C-H BCPs change from a circularly symmetrical distribution (≈ 0 a.u.) of ρ(r) at the PRC-1 and TS-1 to 0.131 a.u. at the PC-1. Additionally, the value of ellipticities remain nearly constant (in the range of 0.026 - 0.031 a.u.) at the O···H BCPs.
At the TS-1, simultaneously a new covalent bond between the O and H atoms appears and the 1,4-pentadiene C-H bond becomes weaker. The nature of the C···H and O···H BCPs bonds at the TS-1 structure is interesting because the proton becomes dicoordinated as is shown by the negative values of ∇2ρ(r) (-0.120 and -0.516 a.u.) and of ρ(r) between 0.1 and 0.2 a.u., respectively. Nevertheless, at this point of the reaction path the C···H bond is weaker. This is evidenced by the values of ∇2ρ(r), which are less negative than for the H···O BCP, which is also in agreement with a "late" transition state as it is common for endothermic reactions. These topological properties calculated at the C···H and H···O BCPs show that these interactions are not purely electrostatic and that a covalent character should be attributed to these bonds.
In closed shell atomic interactions,the electronic charge is concentrated separately in each atomic basin in shell-like regions, similar to those found in an isolated atom or ion. In contrast, in shared shell interactions, the electronic charge is concentrated in the neighborhood of the interatomic surface and extended over both atomic basins which exerts the attractive forces on the nuclei. In such atomic interactions, ∇2ρ(r) is negative,the contraction along the bond trajectory will show a local gain in the potential energy, |V(r)|, due to the increase in the density which is shared by both atoms. This trend is found for the energy densities calculated at the O-H BCPs: the kinetic energy density values, G(r smoothly increase (from 0.021 at PRC- 1, 0.083 at TS-1 to 0.067 a.u. at PC-1) and |V(r)| drastically increases going from the PRC-1 to the PC-1 (from 0.021 at PRC-1, 0.295 at TS-1 to 0.764 a.u. at PC-1). Furthermore, an opposite trend is observed for the values of G(r) and |V(r)| calculated at the C-H BCPs: it is found that G(r) is slightly lowered at the TS-1 and is nearly 50% lower at the PC-1 (0.035 at PRC-1, 0.031 at TS-1 and 0.015 a.u. at PC-1). Notwithstanding,|V(r)| at the TS-1 and PC-1 C-H BCP is remarkably reduced (0.304 at PRC-1, 0.091 at TS-1 and 0.018 a.u. at PC-1). This decrease of |V(r)| at the C-H BCP allows us to explain the weakening of the C-H bond at the TS-1 and the change of nature from a shared shell C-H bond interaction at the PRC-1 to a closed shell hydrogen bond interaction at the PC-1. As a consequence of these trends, important changes of the |V(r)|/G(r) relationship are found. At the PRC-1, the H···O BCP |V(r)|/G(r) = 1 and the C-H BCP |V(r)|/G(r) is 8.6, which indicates closed shell and pure shared shell interactions, respectively The |V(r)|/G(r) ratios larger than 2 (3.0 and 3.6) which are obtained at the TS-1 at the C···H and O···H BCPs, and also the negative values of H(r) (-0.061 and -0.212 a.u.) are further indications of the covalent pure shared shell nature of these bonds.
TOPOLOGICAL ATOMIC PROPERTIES
In order to achieve a deeper understanding of the atomic interactions between fragments in the reaction sites, the individual atomic contributions to the changes (Δ) of v(Ω), M(Ω), q(Ω), Ee(Ω) and V(Ω) were analyzed, see Table 6. Based on the topology of ρ(r), the QTAIM theory defines an atom as a bounded portion of real space, known as atomic basin denoted by Ω. All information about a particular atom is contained in its finite volume and the properties can be obtained by integrating the corresponding property over Ω.
In the bis-allylic C, a significant increase of Δv(Ω) is observed from PRC-1(0.62 a.u.) to the PC-1 by 20.60 a.u.,an indication of the redistribution of the electronic density shown by a more negatively charged carbon atom at the PC-1 (Δq(Ω) = -0.18 a.u.). At the TS- 1, Δv(Ω) has an intermediate value of 12.43 a.u., and Δ|M(Ω)| has the largest increase (0.37 a.u.). In the course of the reaction a minor stabilization of this C atom is found at the TS-1 and PC-1.
The situation changes for the properties of the bis-allylic H and O atoms. At the PRC-1, the H and O atoms experience a decrease of v(Ω) (of -17.01 and -19.10 a.u.) due to the formation of a C-H···O hydrogen bond between the 1,4-pentadiene and the superoxide radical anion. The formation of TS-1 leads to a ≈2-fold decrease of v(Ω) (-33.28 and -36.40 a.u. respectively), and at the PC-1 the v(Ω) of the H and O atoms increases, becoming similar to that of the PRC-1.
On the other hand, the O atom is significantly energetically stabilized (ΔEe(Ω) = -0.17 and ΔV(Ω) = -0.34) at the PC- 1. This result is expected taking into account that these changes of Ee(Ω) and V(Ω) are due to the formation of a more stable hydroperoxyl radical compared to the isolated superoxide radical anion O atom. Moreover, the O atom exhibits a significant reduction of its Δ|M(Ω)|(0.10 a.u.) and a very low increase of q(Ω) at the PC-1. In contrast, the proton becomes more positively charged(Δq(Ω) = 0.42) and experiences a minor increase (0.03 a.u.) of Δ|M(Ω)|at the TS-1. PC-1 shows an important energetic destabilization compared with the isolated 1,4-pentadiene H atom energies (at the TS-1 ΔEe(Ω) = 0.23 and ΔV(Ω) = 0.47 and at PC-1 (ΔEe(Ω) = 0.20 and ΔV(Ω) = 0.77 a.u.). In other words, the energetic destabilization of the TS-1 and PC-1 is mostly due to the slight increase in the energy of the bis-allylic dicoordinated proton at these complexes, which is compensated by an energetical stabilization of the superoxide anion O atom.
Characterization of the O-H···C hydrogen bonds in the product complex
A strong O-H···C hydrogen bond appears in the PC-1, which is characterized by a stabilization energy of 81.6 kJ/ mol, as noted before. An understanding of the nature of this hydrogen bond can be achieved by inspecting the local and atomic properties at the hydroperoxyl radical proton. ρ(r) at the bond critical point is a key descriptor of hydrogen bonds. The H···C BCP has values of ρ(r) and ∇2ρ(r) of 0.030 and ≈ 0.05 a.u., a ratio |V(r)|/G(r) = 1.2 and a slightly negative value of H(r). These topological properties of the electron density at the H···C BCP are typical for transit closed shell interactions (∇2ρ(r) > 0) (30-31). Furthermore, the values of |V(r)|/G(r) and H(r) lie at the boundary between a pure closed shell and a transit closed shell interaction.
Furthermore, the four criteria of Popelier (38) for hydrogen bonding involving integrated topological atomic properties include the following: (1) energetic destabilization, (2) loss of q(Ω),(3) reduced v(Ω), and (4) decrease of |M(Ω)| of the hydrogen atom, compared to a non-hydrogen-bonded H atom reference specie. The changes in the atomic properties (Δ) in v(Ω), M(Ω), q(Ω) and Ee(Ω) calculated relative to the hydroperoxyl radical H atom are shown in parentheses in Table 6. In agreement with these criteria, the O-H···C hydrogen bond of PC-1 exhibits a large loss of v(Ω) of 10.64 a.u., of |M(Ω)| of 0.04, of q(Ω) of 0.02 and an energetic destabilization of 0.01 a.u.
CONCLUSIONS
The protonation of superoxide radical anion by 1,4-pentadiene was investigated by means of the density functional theory (DFT). A number of geometrical,thermochemical and topological parameters were computed at the B3LYP/6- 311+G(3df,2p) theoretical level. Moreover, the molecular wave functions of the intermediates as well as the effects of hydrogen bonding involved on the reaction mechanism were analyzed in the framework of the Quantum Theory of Atoms in Molecules (QTAIM) approach.
We found four bis-allylic proton transfer reaction pathways and the formation of strong hydrogen bonded complexes between the 1,4-pentadien-3-yl anion and the hydroperoxyl radical as the reaction products. These product complexes (PCs) are more stable than the isolated reactants by -28.9 and -23.1 kJ/mol. The reaction proceeds via pre-reactive complexes (PRCs) which are more stable than the PCs (11.5 - 25.3 kJ/mol) and the transition states (TSs) (14.4 - 25.5 kJ/mol). Moreover, taking into account the very small energy difference between the PCs and the TSs (1.5 - 5.8 kJ/mol), it can be expected that the reverse reaction to the PRCs competes with the direct reaction. Nevertheless, due to the strong binding energy of the PCs (-81.6 to -86.1 kJ/mol) it is predicted that the backward reaction, which leads to the PRCs, and the forward reaction, which releases the free products (1,4-pentadien-3-yl anion and the hydroperoxyl radical), is not plausible. This is also in agreement with the fact that the net proton transfer reaction leading to the free products is endothermic (43.3 - 54.0 kJ/mol) and non-spontaneous (44.2 - 48.5 kJ/mol).
A more detailed understanding of the mechanism of the proton transfer reaction from 1,4-pentadiene to superoxide anion is obtained by analyzing the topological local and atomic properties of the electron density distribution at the bisallylic C-H and O-H bond critical points at the pre-reactive, transition states and product complexes. The rupture of the bis-allylic 1,4-pentadiene C-H bond at the product complexes and the appearan ce of the new O-H bond is confirmed by the change in the electron density (ρ(r)) and its Laplacian (∇2ρ(r)) at each bond critical point (BCP). On the one hand, a depletion of ρ(r) at the bis-allylic C BCPs when going from the pre-reactive complexes to the product complexes is observed (0.269 to 0.030 a.u.). On the other hand, the newly formed O-H bond undergoes a transition from a C-H···O hydrogen bond in the pre-reactive complexes to a covalent O-H bond in the product complexes evidenced by an increase of ρ(r) from 0.029 to 0.340 a.u., and by the change of sign of the ∇2ρ(r) from positive to negative. Moreover, at the transition states a dicoordinated character of the bis-allylic proton at the transition states is obtained as is shown by negative values of ∇2ρ(r) and of the total energy density (H(r)) and also by |V(r)|/G(r) values larger than two. The analysis of these parameters suggests that the bis-allylic proton of 1,4-pentadiene is connected to the carbon atom and to the superoxide radical anion oxygen atom by a pure shared shell covalent interaction.
At the hydrogen bonded product complexes between hydroperoxyl radical and 1,4-pentadien-3-yl anion, a strong O-H···C hydrogen bond appears. It was found that the topological local properties of the electron density at the H···C bond critical points are typical for non-covalent transit closed shell interactions (∇2ρ(r) > 0). The values of |V(r)|/G(r) and H(r)lie at the boundary between a pure closed shell and a transit closed shell interaction. Furthermore, energetic destabilization (Ee(Ω)), a loss of charge (q(Ω)), and a decrease of dipolar polarization (M(Ω)) and of volume (v(Ω)) of the hydroperoxyl radical hydrogen atom are observed.
ACKNOWLEDGMENTS
The present work received financial support from Fundación para la Promoción de la Ciencia y la Tecnología del Banco de la República (2630 Project) and Universidad Industrial de Santander (5169 Project). A. Rodríguez-Serrano wishes to thank Colciencias ’Programa Generación Bicentenario, becas Francisco José de Caldas’.
REFERENCES
1. Keesee, R. G.; Lee, N. J.; Castleman Jr., A. W. Atmospheric negative ion hydration derived from laboratory results and comparison to rocket-borne measurements in the lower ionosphere. J. Geophys. Res. 1979. 84: 3719-3722. [ Links ]
2. Fahey, W. D.; Böhringer, H.; Fehsenfeld, F. C.; Ferguson, E. E. Reaction rate constants for O2 -(H2O)n ions n = 0-4 with O3, NO, SO2 and CO2. J. Chem. Phys. 1982. 76: 1799-1805. [ Links ]
3. Diaz-Uribe, C. E.; Daza, M. C.; Martinez, F.; Páez-Mozo, E. A.; Guedes, C. L. B.; Di Mauro, E. Visible light superoxide radical anion generation by tetra(4-carboxyphenyl) porphyrin/TiO2: EPR characterization. J. Photochem. Photobiol. A: Chem. 2010. 215: 172-178. [ Links ]
4. Schöneich, C. Reactive oxygen species and biological aging: a mechanistic approach. Exp. Gerontol. 1999. 34: 19-34. [ Links ]
5. Salvemini, D.; Wang, Z. -Q.; Zweier, J. L.; Samouilov, A.; Ma carthur, H.; Misko, T. P.; Currie, M. G.; Cuzzocrea, S.; Sikorski, J. A.; Riley, D. P. A nonpeptidyl mimic of superoxide dismutase with therapeutic activity in rats. Science 1999. 286: 304-306. [ Links ]
6. Jonson, J. E.; Isaksen, I. S. A. Tropospheric ozone chemistry: the impact of cloud chemistry. J. Atmos. Chem. 1993. 16: 99-122. [ Links ]
7. Babior, B. M. NADPH oxidase: an update. Blood 1999. 93: 1464-1476. [ Links ]
8. Brand, M. D.; Affourtit, C.; Esteves, T. C.; Green, K.; Lambert, A. J.; Miwa, S.; Pakay, J.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of un coupling proteins. Free Radic. Biol. Med. 2004. 37: 755-767. [ Links ]
9. Afanas'ev, I. Superoxide Ion Chemistry and Biological Implications. USA, CRC Press. 1989. [ Links ]
10. Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005. 120: 483-495. [ Links ]
11. Bielski, B. H.; Arudi, R. L.; Sutherland, M.W. A study of the reactivity of 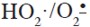 with unsaturated fatty acids. J. Biol. Chem. 1983. 258: 4759-4761. [ Links ]
with unsaturated fatty acids. J. Biol. Chem. 1983. 258: 4759-4761. [ Links ]
12. Sawyer, D. T.; Valentine, J. S. How super is superoxide?. Acc. Chem. Res. 1981. 14: 393-400. [ Links ]
13. Stanley, J. P. Reactions of superoxide with peroxides. J. Org. Chem. 1980. 45: 1413-1418. [ Links ]
14.Starý, J.; Hladký, E.Systematic study of the solvent extraction of metal ß-diketonates.Anal. Chim. Acta1963. 28:227-235. [ Links ]
15. Durairaj, R. B. Resorcinol: chemistry, technology, and applications. Berlin, Springer Verlag Gmbh & Co. Kg. 2005. [ Links ]
16. Frimer, A. A.; Gilinsky-Sharon, P. Reaction of superoxide anion radical [O2 -.] with cyclohex-2-en-1-ones. Tetrahedron Lett. 1979. 44: 4331- 4334. [ Links ]
17. Gibian, M. J.; Sawyer, D. T.; Ungermann, T.; Tangpoonpholvivat, R.; Morrison, M. M. Reactivity of superoxide ion with carbonyl compounds in aprotic solvents. J. Am Chem. Soc. 1979. 101: 640-644. [ Links ]
18. DiBiase, S. A.; Wolak Jr., R. P.; Dishong, D. M.; Gokel, G. W. Crown cation complex effects. 10. Potassium tert-Butoxide mediated penultimate oxidative hydrolysis of nitriles. J. Org. Chem. 1980. 45: 3630-3633. [ Links ]
19. Bell, A. J.; Wright, T. G. Experimental and theoretical studies on the complex formed between H2S and 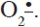 J. Phys. Chem. A 2004. 108: 10486-10490. [ Links ]
J. Phys. Chem. A 2004. 108: 10486-10490. [ Links ]
20. Dhaouadi, Z.; Nsangou, M.; Garrab, N.; Anouar, E. H.; Marakchi, K.; Lahmar, S. DFT study of the reaction of quercetin with 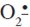 and -OH radicals. J. Mol. Struct. (Theochem) 2009. 904: 35-42. [ Links ]
and -OH radicals. J. Mol. Struct. (Theochem) 2009. 904: 35-42. [ Links ]
21. Cardey, B.; Foley, S.; Enescu, M. Mechanism of thiol oxidation by the superoxide radical. J. Phys. Chem. A 2007. 111: 3046-13052. [ Links ]
22. Cardey, B.; Enescu, M. Cysteine oxidation by the superoxide radical: A theoretical study. ChemPhysChem 2009. 10: 1642-1648. [ Links ]
23. Durand, G.; Choteau, F.; Pucci, B.; Villamena, F. A. Reactivity of superoxide radical anion and hydroperoxyl radical with alpha-phenyl-ntert- butylnitrone (PBN) derivatives. J. Phys. Chem. A 2008. 112: 12498- 12509. [ Links ]
24. Villamena, F. A.; Locigno, E. J.; Rockenbauer, A.; Hadad, C. M.; Zweier, J. L. Theoretical and experimental studies of the spin trapping of inorganic radicals by 5,5-dimethyl-1-pyrroline n-oxide (DMPO). 1. Carbon dioxide radical anion. J. Phys. Chem. A 2006. 110: 13253-13258. [ Links ]
25. Villamena, F. A.; Xia, S.; Merle, J. K.; Lauricella, B.; Tuccio, R.; Hadad, C. M.; Zweier, J. L. Role of intramolecular H-bond and electrostatics on the reactivity of superoxide radical anion with cyclic nitrones. J. Am. Chem. Soc. 2007. 129: 8177- 8191. [ Links ]
26. Field, S. M.; Villamena, F. A. Theoretical and experimental studies of tyrosyl hydroperoxide formation in the presence of H-bond donors. Chem. Res. Toxicol. 2008. 21: 1923-1932. [ Links ]
27. Villamena, F. A.; Rockenbauer, A.; Gallucci, J.; Velayutham, M.; Hadad, C. M.; Zweier, J. L. Spin trapping by 5-carbamoyl-5-methyl- 1-pyrroline N-oxide (AMPO): Theoretical and Experimental Studies. J. Org. Chem. 2004. 69: 7994-8004. [ Links ]
28. Han, Y.; Tuccio, B.; Lauricella, R.; Villamena, F. A. Improved spin trapping properties by beta-cyclodextrin- cyclic nitrone conjugate. J. Org. Chem. 2008. 73: 7108-7117. [ Links ]
29. Burgett, R. A.; Bao, X.; Villamena,F. A. Superoxide radical anion adduct of 5,5-dimethyl-1-pyrroline N-oxide (DMPO). 3. Effect of mildly acidic pH on the thermodynamics and kinetics of adduct formation. J. Phys. Chem. A 2008. 112:2447-2455. [ Links ]
30. Zárate, X.; Daza, M. C.; Villaveces, J. L. Hydrogen bonds CH ···O in superoxide anion radical - 1,4-pentadiene complexes. J. Mol. Struct. (Theochem) 2009. 893: 77- 83. [ Links ]
31. Ernst, R. D. Structure and bonding: Vol. 57, Berlin, Springer Verlag. 1984. [ Links ]
32. Powell, P. Acyclic Pentadienyl metal complexes in: Advances in organometallic chemistry Vol. 26. New York, Academic Press Inc. 1986. [ Links ]
33. Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993. 98: 5648-5652. [ Links ]
34. Lee, C.; Yang, W.; Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988. 37: 785-789. [ Links ]
35. Gaussian 03, Revision B.05, Gaussian, Inc., Pittsburgh, PA, USA, 2003. [ Links ]
36. Boys, S. F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970. 19: 553-566. [ Links ]
37. Bader, R. F. W. Atoms in Molecules: A Quantum Theory. Oxford, Oxford University Press. 1990 (Reprinted 2000). [ Links ]
38. Bader, W.; Essen, H. The characterization of atomic interactions. J. Chem. Phys. 1984. 80: 1943-1960. [ Links ]
39. Koch, U.; Popelier, P. L. A. Characterization of C-H-O hydrogen bonds on the basis of the charge density. J. Phys. Chem. 1995. 99: 9747-9754. [ Links ]
40. Popelier, P. L. A. Characterization of a dihydrogen bond on the basis of the electron density J. Phys. Chem. A 1998. 102: 1873-1878. [ Links ]
41. Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. J. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X- Hâ¯F-Y systems. J. Chem. Phys. 2002. 117: 5529-5542. [ Links ]
42. Biegler-König, F.; Schönbohm, J.; Bayles, D. J. AIM2000--A Program to analyze and visualize Atoms in Molecules. J. Comput. Chem. 2001. 22: 545-559. [ Links ]
43. Reed, A. E.; Weinstock, R. B.; Weinhold, F. A. Natural Population Analysis. J. Chem. Phys. 1985. 83: 735-746. [ Links ]
44. Reed, A. E.; Curtiss, L. A.; Weinhold, F. Intermolecular interactions from a Natural Bond Orbital, donoracceptor viewpoint. Chem. Rev. 1988. 88: 899-926. [ Links ]
45. Glendening, E. D.; Badenhoop, J. K.; Reed, A. E.; Carpenter, J. E.; Bohmann, J. A.; Morales, C. M.; Weinhold, F. NBO 5.0, Theoretical Chemistry Institute, Madison, University of Wisconsin. 2001. [ Links ]
46. Szori, M.; Viskolcz, B. Conformation analysis of 1,4-pentadien- 3-yl radicals by ab initio and DFT methods .J. Mol. Struct. (Theochem) 2003. 666-667: 153-158. [ Links ]
47. MacInnes, I.; Walton, J. C. Rotational barriers in pentadienyl and pent- 2-en-4-ynyl radicals. J. Chem. Soc., Perkin Trans. 1985. 2: 1073-1076. [ Links ]
48. Davies, A. G.; Griller, D.; Ingold, K. U.; Lindsay, D. A.; Walton, J. C. An electron spin resonance study of pentadienyl and related radicals: homolytic fission of cyclobut-2-enylmethyl radicals. J. Chem. Soc., Perkin Trans. 1981. 2: 633-641. [ Links ]
49. Brady Clark, K.; Culshaw, P. N.; Griller, D.; Lossing, F. P.; Mart inho Simões, J. A.; Walton, J. C. Studies of the formation and stability of pentadienyl and 3-substituted pentadienyl radicals. J. Org. Chem. 1991. 56: 5535-5539. [ Links ]
50. Berkowitz, J.; Ellison, G. B.; Gutman, D. Three methods to measure RH bond energies. J. Phys. Chem. 1994. 98: 2744-2765. [ Links ]
51. McMillen, D. F.; Golden, D. M. Hydrocarbon bond dissociation energies. Ann. Rev. Phys. Chem. 1982. 33: 493-532. [ Links ]
52. de Heer, M. I.; Korth, H. -G; Mulder, P. Polymethoxy phenols in solution: O-H bond dissociation enthalpies, structures, and hydrogen bonding. J. Org. Chem. 1999. 64: 6969-6975. [ Links ]
53. Hammond, G. S. A correlation of reaction rates. J. Am. Chem. Soc. 1955. 77: 334-338. [ Links ]
