










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Química
Print version ISSN 0120-2804
Rev.Colomb.Quim. vol.42 no.1 Bogotá Jan./Apr. 2013
Método acoplado Autodock–PM6 para seleccionar la mejor pose en estudios de acoplamiento molecular
Autodock–PM6 method to choose the better pose in molecular docking studies
A Autodock–PM6 metodo pra seleccionar o melhor pose en estudios de acoplamiento molecular
Margarita Velásquez1, Juan Drosos1, Carlos Gueto1, Johana Márquez1, Ricardo Vivas–Reyes1**
1 Departamento de Química, Facultad de Ciencias, Universidad Nacional de Colombia, sede Bogotá, Crr. 30 No. 45–03, Bogotá DC, Colombia. Grupo de investigación Estado Sólido y Catálisis Ambiental (Esca).
*Autor de Correspondencia. Email: rvivasr@unicartagena.edu.co
Recibido: 15 de enero de 2013 • Aceptado: 29 de abril de 2013
Resumen
El acoplamiento molecular (conocido como docking) es una técnica de mecánica molecular ampliamente utilizada para predecir energías y modos de enlace entre ligandos y proteínas, información de gran utilidad en el estudio de nuevos compuestos con efectos terapéuticos. No obstante, los resultados obtenidos mediante esta técnica tienden a la subjetividad, debido a que los programas utilizados para llevarla a cabo proporcionan más de un criterio de selección de la mejor pose. En la presente investigación se aplicó el método semiempírico PM6 a los resultados del acoplamiento, obteniendo con ello mejorías en el proceso de selección de la mejor pose pues se obtuvieron poses con alta probabilidad de unión al sitio activo de su receptor y con energías de unión menores a las reportadas por los criterios de selección ofrecidos por el programa de docking.
Palabras Clave: acoplamiento molecular, método semiempírico PM6, autodock, pose.
Abstract
The acoplamiento molecular (conocido como docking) es una técnica de mecánica molecular ampliamente utilizada para predecir energías y modos de enlace entre ligandos y proteínas, lo que proporciona información de gran utilidad para el estudio de nuevos compuestos con efectos terapéuticos. No obstante, los resultados obtenidos mediante esta técnica tienden a la subjetividad, debido a que los programas utilizados para llevarla a cabo proporcionan más de un criterio de selección de la mejor pose. En la presente investigación, se aplicó el método semiempírico PM6 a los resultados del acoplamiento, obteniendo con ello mejorías en el proceso de selección de la mejor pose al observarse finalmente poses con alta probabilidad de unión al sitio activo de su receptor y con energías de unión menores a las reportadas por los criterios de selección ofrecidos por el programa de docking.
Keywords: Molecular Coupling, PM6 semi-empirical method, AutoDock, Pose.
Resumo
El acoplamiento molecular (conocido como docking) es una técnica de mecánica molecular ampliamente utilizada para predecir energías y modos de enlace entre ligandos y proteínas, lo que proporciona información de gran utilidad para el estudio de nuevos compuestos con efectos terapéuticos. No obstante, los resultados obtenidos mediante esta técnica tienden a la subjetividad, debido a que los programas utilizados para llevarla a cabo proporcionan más de un criterio de selección de la mejor pose. En la presente investigación, se aplicó el método semiempírico PM6 a los resultados del acoplamiento, obteniendo con ello mejorías en el proceso de selección de la mejor pose al observarse finalmente poses con alta probabilidad de unión al sitio activo de su receptor y con energías de unión menores a las reportadas por los criterios de selección ofrecidos por el programa de docking.
Palavras-chave: acoplamiento molecular, método semiempírico PM6, autodock, pose.
Introducción
Anteriormente, la búsqueda de nuevos compuestos con efectos terapéuticos implicaba la síntesis a través de varias rutas de los supuestos compuestos con la propiedad buscada o el cribado de productos naturales, esfuerzos que pueden ser categorizados como exploraciones aleatorias o al azar, en lugar de orientaciones racionales. En los últimos años se han dado a conocer una serie de grandes avances en los cálculos teóricos que han favorecido el uso y la popularidad de los estudios in silico (computacionales) para el análisis de moléculas de interés biológico; ello se ha convertido en parte integral de la investigación industrial y académica dirigida al diseño y descubrimiento de fármacos (1).
Las herramientas computacionales han evolucionando de tal forma que se han transformando en tecnologías cada vez más importantes para la búsqueda de moléculas candidatas a fármacos, mediante la se lección de moléculas denominadas 'cabezas de serie' o leads a partir de bases de datos. Entre estas herramientas se encuentra el modelado molecular, técnica que permite obtener moléculas –reales o virtuales– que poseen gran probabilidad de exhibir una acción específica y que hace posible, incluso, predecir su biodisponibilidad y toxicidad (1, 2).
A su vez, dentro de las herramientas de modelado molecular se encuentra el acoplamiento molecular (docking), técnica de gran utilidad para predecir la estructura de los complejos intermoleculares que se establecen entre dos o más moléculas. Uno de los casos más estudiados es la interacción proteína–ligando, cuyo objetivo principal es predecir energías y modos de enlace, y que suele hacerse previamente a un estudio experimental (3, 6).
En estudios donde se aplica el acoplamiento molecular, usualmente se considera la mejor pose aquella que reporta la menor energía de unión. Alternativamente, puede ser seleccionada del cluster más poblado entre los clusters obtenidos después del acoplamiento molecular y, en ocasiones, también se selecciona el menor valor de RMSD (Root Mean Square Deviation) con respecto a una estructura de referencia (4, 7).
Aunque los parámetros energéticos son muy importantes al momento de efectuar esta selección, es conveniente tener en cuenta que los campos de fuerza empleados por los métodos de mecánica molecular (MM) (entre éstos, el acoplamiento molecular) llevan consigo algunas aproximaciones que lógicamente conducirían a resultados no tan reales. Estas aproximaciones se relacionan principalmente con el hecho de considerar sólo la disposición nuclear de los átomos, prescindiendo de los electrones, al asumir que éstos se distribuirán óptimamente a su alrededor.
Es evidente que, a pesar del éxito alcanzado por los programas de docking, es necesario alcanzar un mayor rendimiento y confiabilidad de estos métodos, razón por la cual en esta contribución se plantea un método que arroje resultados de la simulación de la interacción más próximos a la realidad.Persiguiendo este objetivo, en un estudio previo (8) realizado por nuestro grupo de investigación, se utilizó el método semiempírico de mecánica cuántica PM6 (9, 10) con el cual se recuperaría de los estudios de docking molecular la información perdida en términos de interacciones y energía de acoplamiento. Para tal estudio, la metodología arrojó resultados satisfactorios (9).
Este hecho se utilizó como punto de partida para el desarrollo del presente artículo, en el cual se evaluó la efectividad que tiene la aplicación del método semiempírico PM6 como complemento a los estudios de docking, utilizando para ello un grupo de complejos pertenecientes a la serie Astex Diverse (11) diseñada para mejorar el rendimiento y la exactitud de los programas de docking y que se distingue por contener estructuras modernas (archivos PDB depositados después del 11 de agosto del 2000) con resolución de 2.5 Å o menor y con ligandos similares a fármacos.
Finalmente, el uso de esta metodología acoplada permite obtener, poses con alta probabilidad de unión al sitio activo de su receptor para cada ligando y con energías de unión menores a las reportadas por los criterios de selección ofrecidos por el programa de docking.
Materiales y métodos
Selección de los complejos proteína–ligando
Para el desarrollo de este trabajo se utilizaron 22 complejos proteína–ligando (Tabla 1) tomados de la base de datos Protein Data Bank (PDB) (12) y seleccionados de la serie de prueba Astex Diverse (11). Esta serie está constituida por 85 complejos proteína–ligando que cumplen principalmente con los siguientes criterios: las dianas incluidas son estructuralmente diversas, aproximadamente el 90% de estas son utilizadas como blancos directos en proyectos de descubrimiento de fármacos y el resto están dentro de las llamadas moléculas off target, entre las cuales se encuentran los citocromos P450. Por otra parte, la mayoría de ligandos clasifican, ya sea como drogas aprobadas, en etapa de ensayos clínicos, o compuestos obtenidos en proyectos de descubrimiento de fármacos. Por último, se utilizaron estructuras de alta calidad para la cuales la densidad electrónica del ligando es compatible con el modo de unión (11).
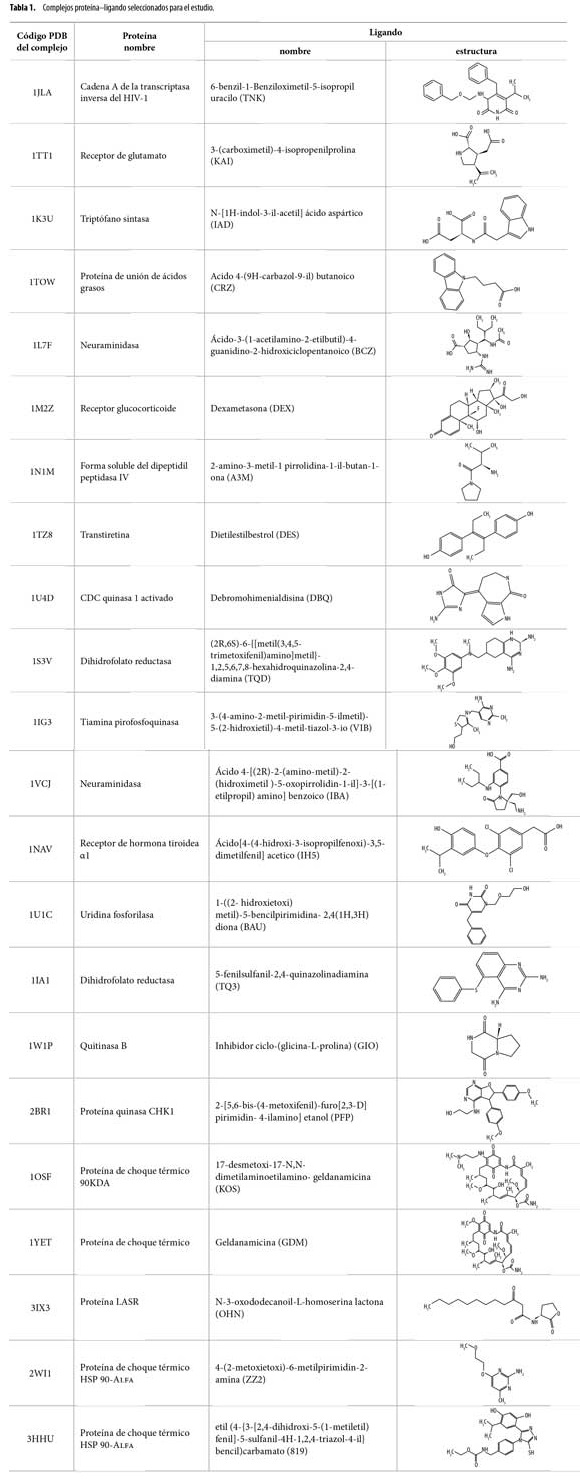
Evaluación del programa de docking
Para el acoplamiento se utilizó el programa AutoDock v4.2 (13, 14). Se realizó un docking ciego entre cada uno de los 22 complejos proteína– ligando y su respectivo receptor, solicitándose para tal fin la generación de 100 poses en cada caso.
Posteriormente, se realizó una inspección visual de los resultados en el programa AutoDockTools v1.5.4 (15) (ADT), a fin de observar el grado de similitud existente entre las poses generadas y las poses nativas.
Preparación de receptores y ligandos
La preparación de los receptores y ligandos se llevó a cabo utilizando el programa Discovery Studio 2.5 (16).
El tratamiento de los receptores consistió en la extracción del ligando y la eliminación de moléculas de agua y cofactores con los cuales vienen resueltas sus estructuras cristalinas.
Por su parte, el tratamiento de los ligandos se basó en la revisión de las posiciones de sus átomos y de todos sus enlaces, tomando como referencia la estructura 2D mostrada en la base de datos PDB. En este punto también fueron adicionados los átomos de hidrógeno a las estructuras.
Acoplamiento molecular
El acoplamiento se realizó bajo los siguientes parámetros: libre rotación de todos los enlaces de los ligandos con libertad conformacional, adición de cargas de Gasteiger a la proteína y al ligando, adición de hidrógenos a la proteína, generación de 100 poses para cada ligando y utilización del Algoritmo Genético Lamarckiano (LGA). Este último se utilizó bajo las siguientes condiciones: 100 corridas del GA, 150 tamaño de población, 2 500 000 máximo número de evaluaciones, 27 000 máximo número de generaciones, 0.02 tasa de mutación del gen y 0.08 rata de cruza.
Selección de las mejores poses
Para seleccionar la mejor pose para cada ligando se utilizaron los siguientes criterios:
Criterio 1. Energía de unión del complejo: se toma como mejor pose aquella que reporta la menor energía de unión entre todas las poses obtenidas.
Criterio 2. Distribución de energía (cluster): se toma como mejor pose aquella que ocupa la primera posición dentro del clúster más poblado.
Recategorización de los resultados del docking utilizando el método semiempírico PM6
Para cada proteína se seleccionó una cavidad de 5 Å con centro en el sitio activo. Cada cavidad y su ligando se optimizaron utilizando el método semiempírico PM6, incluido en el paquete de métodos semiem píricos MOPAC2009 (17). Las estructuras optimizadas y los archivo de salida arrojados al usar el programa AutoDock v4.2 se utilizaron como punto de partida para el proceso de recategorización de las poses obtenidas del acoplamiento, mediante la reoptimización de todas las geometrías obtenidas.
Acoplamiento molecular
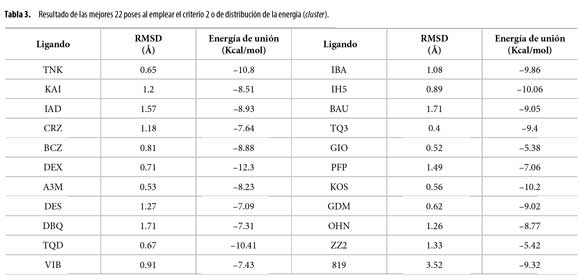
Los resultados obtenidos de los cálculos de acoplamiento molecular fueron categorizados de acuerdo con los criterios 1 y 2, previamente mencionados (Tablas 2 y 3).
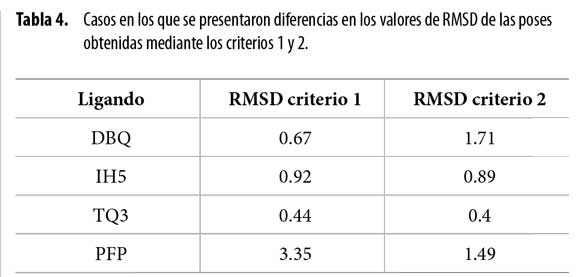
. Este porcentaje está representado por los ligandos DBQ, IH5, TQ3 y PFP, para los cuales la mejor pose seleccionada no coincidió con la pose obtenida mediante el criterio1 (Tabla 4).
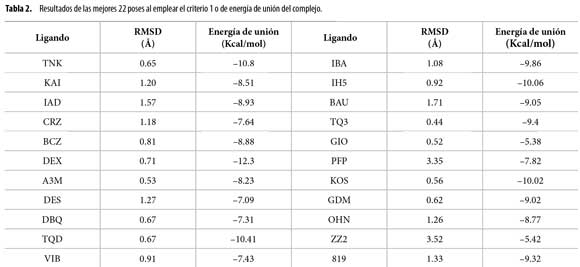
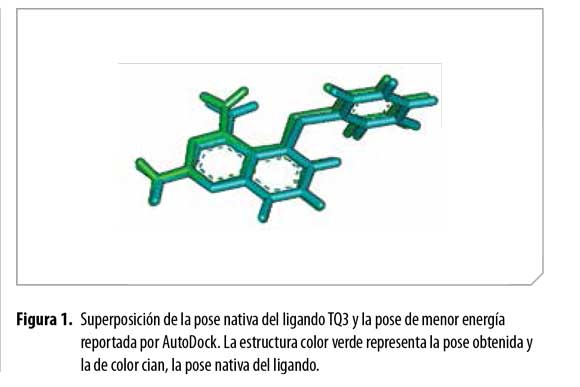
Al observar en detalle y comparar –usando el programa AutoDockTools (ADT)–, la pose nativa vs. la pose obtenida para cada ligando, se observó gran similitud conformacional entre éstas para la mayor parte de los casos, lo cual fue respaldado por los valores de RMSD(Root Mean Square Deviation) arrojados para dichas poses (Tablas 2 y 3 y Figura 1).
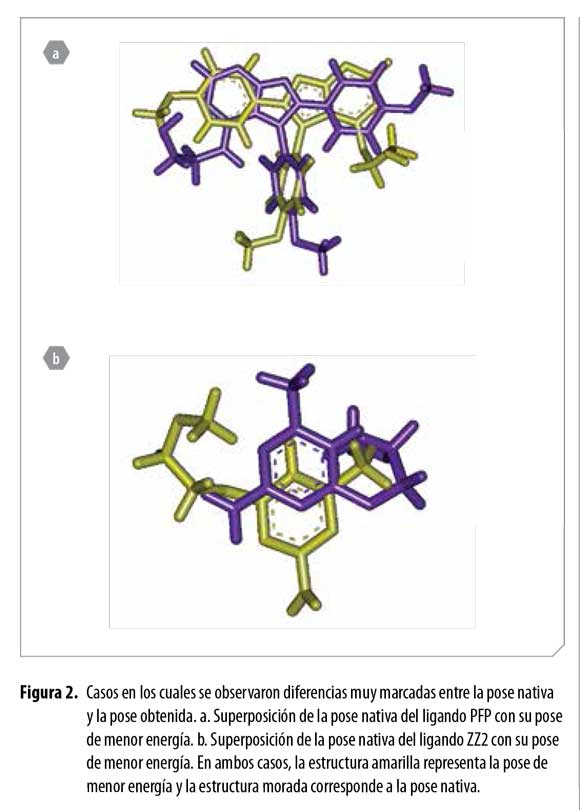
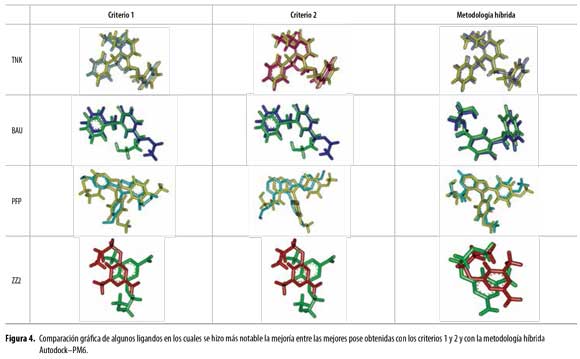
No obstante, se encontraron casos particulares en los cuales no se observó dicha similitud (ligandos PFP y ZZ2) (Figura 2 y Tablas 2 y 3). En estos casos, las orientaciones espaciales de los átomos de la mejor pose difieren notablemente de los átomos de la pose nativa, hecho que lleva a pensar que este tipo de criterio en cierta medida no ofrece una selección 100% confiable.
Al aplicar el criterio de distribución de la energía (cluster) (Tabla 3) y comparar, se hizo evidente que aproximadamente en el 82% de los casos las poses coincidían con las obtenidas mediante el criterio 1 (Tabla 2), lo que era de esperarse ya que, generalmente, en el cluster más poblado se encuentran agrupadas las poses de menor energía. Sin embargo, es necesario fijarse en el 18% de los casos restantes en los cuales no se presentaron coincidencias
Al visualizar ambas poses con ADT (tanto las obtenidas por el criterio 1, como las obtenidas por el criterio 2), se notó que en la mayor parte de los casos existe gran similitud conformacional entre éstas y su pose nativa correspondiente, excepto para el ligando PFP con el criterio 1.
Teniendo en cuenta lo anterior, se puede afirmar que no existe concordancia total entre los datos arrojados por los criterios 1 y 2, corroborándose así la subjetividad que brinda AutoDock para seleccionar las mejores poses en un estudio determinado, dejando a consideración del investigador el criterio a utilizar.
El método semiempírico PM6 se utilizó para optimizar todos los átomos de hidrógeno de los 22 sistemas ligando–proteína estudiados. Debido a que con este método se aporta un enfoque mecánico–cuántico, se obtuvo un reordenamiento más riguroso de los resultados del docking (Tabla 5).
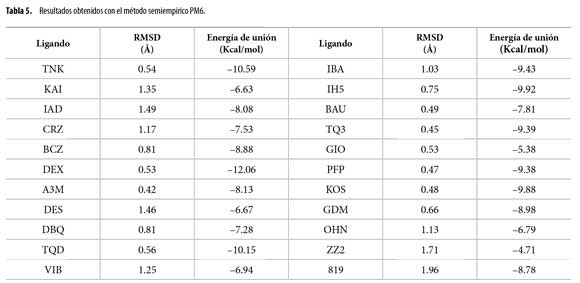
Es evidente que todos los valores de RMSD para las poses obtenidas después de la recategorización con PM6 están por debajo del límite de tolerancia (2 Å), lo cual indica de que esta metodología es capaz de seleccionar como mejor pose aquella que presenta gran semejanza con la pose nativa de aquellos ligandos evaluados dentro de sus receptores biológicos, evitando con esto la subjetivad de los criterios 1 y 2.
Los valores de RMSD mostrados en la Tabla 5 difieren en un 95% con respecto a los de las Tablas 2 y 3. Sin embargo, para el ligando BCZ se reportaron los mismos valores de RMSD (0.81Å), tanto para los criterios 1 y 2, como después de la recategorización con PM6, lo que se encuentra dentro de las posibilidades debido a que este valor de RMSD significa que la mejor pose es muy parecida a la pose nativa.
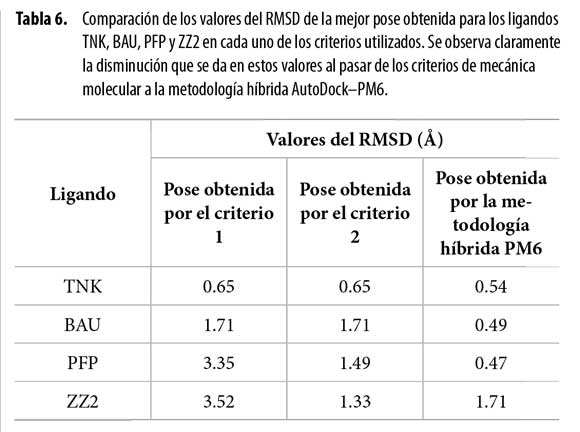
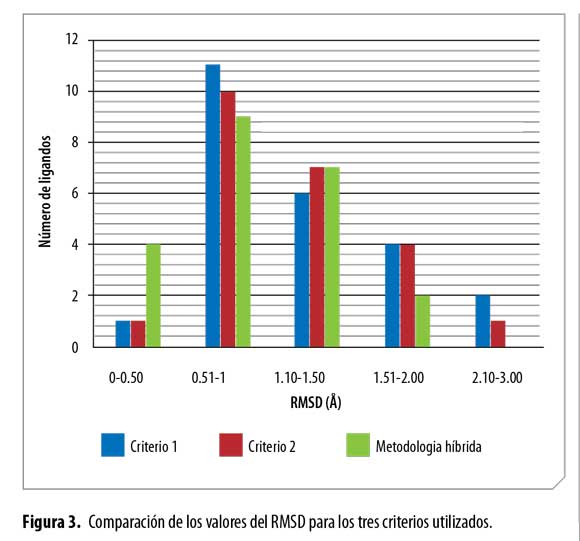
Por otra parte, los valores de RMSD menores que 2 se evidenciaron en un 91% para el criterio 1, en un 95% para el criterio 2 y en un 100% para la metodología híbrida PM6, lo que muestra la superioridad ofrecida por esta última. La mejoría observada en esta metodología sobre los criterios 1 y 2 se ve especialmente reflejada en el caso de los ligandos TNK, BAU, PFP y ZZ2 (Figuras 3 y 4; Tabla 6).
En las Figuras 3 y 4 se muestra de forma global la tendencia observada en los valores RMSD para los tres criterios utilizados, así como su comparación gráfica correspondiente.
A pesar de que AutoDock ofrece un buen sistema de búsqueda conformacional, sus requerimientos impiden considerar la influencia de todos los hidrógenos del sistema ligando–proteína, lo que conduce a una importante pérdida de información representada en términos de interacciones.
Así, la utilización del método semiempírico PM6 para el reordenamiento o recategorización de los resultados del docking favorece en gran medida la selección de los mejores resultados, gracias a que con esta metodología se optimizan todos los hidrógenos que participan en la interacción ligando–receptor, lo que permite lograr una aproximación más real del acoplamiento. Lo anterior se ve confirmado en el hecho que las poses obtenidas del docking con altos valores de RMSD, una vez se optimizan con PM6, reportan valores mucho menores, evidenciando con ello la marcada influencia que posee la consideración de todos los hidrógenos durante el proceso de acoplamiento.
Por otra parte, las mejores poses obtenidas con la metodología híbrida PM6 presentaron valores de RMSD mejores que los ofrecidos por AutoDock, ya que en todos los casos con esta metodología todos valores permanecieron dentro de su límite de tolerancia, hecho que no se observó en los resultados con AutoDock.
De manera general se puede afirmar que la combinación de diferentes técnicas computacionales podrían permitir realizar estudios más rigurosos y, por tanto, más reales a nivel molecular.
Agradecimientos
La realización de este trabajo fue llevada a cabo por el Grupo de Investigación de Química Cuántica y Teórica de la Universidad de Cartagena, gracias al apoyo del Departamento Administrativo de Ciencia, Tecnología e Innovación –Colciencias–, a través del beneficio de la beca-pasantía otorgada mediante el programa Jóvenes Investigadores e Innovadores año 2011.
Referencias
1. Reddy, A.S. , et al. , Virtual screening in drug discovery - a computational perspective. Curr Protein Pept Sci, 2007. 8(4): p. 329-51. [ Links ]
2. Ghersi, D. and R. Sanchez, Improving accuracy and efficiency of blind protein-ligand docking by focusing on predicted binding sites. Proteins, 2009. 74(2): p. 417-24. [ Links ]
3. Zúñiga, A.J.P. and A.R. Domínguez, Simulación del reconocimiento entre proteínas y moléculas orgánicas o docking. Aplicación al diseño de fármacos. Mensaje Bioquímico, 2002. 26. [ Links ]
4. Schulz-Gasch, T. and M. Stahl, Scoring functions for protein–ligand interactions: a critical perspective. Drug Discovery Today: Technologies, 2004. 1(3): p. 231-239. [ Links ]
5. O'Brien, S.E. , et al. , Computational tools for the analysis and visualization of multiple protein-ligand complexes. J Mol Graph Model, 2005. 24(3): p. 186-94. [ Links ]
6. Shoichet, B.K. , et al. , Lead discovery using molecular docking. Curr Opin Chem Biol, 2002. 6(4): p. 439-46. [ Links ]
7. Wong, C.F. , Flexible ligand-flexible protein docking in protein kinase systems. Biochim Biophys Acta, 2008. 1784(1): p. 244-51. [ Links ]
8. Lázaro, J.M. , Evaluación del desempeño de diferentes aproximaciones 3D QSAR. Caso modelo: antitumorales análogos de la geldanamicina con la proteína Hsp90, in Programa de Química. Grupo de Química Cuántica y Teórica2010, Universidad de Cartagena. [ Links ]
9. Bikadi, Z. and E. Hazai, Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminform, 2009. 1: p. 15. [ Links ]
10. Korth, M. , Third-Generation Hydrogen-Bonding Corrections for Semiempirical QM Methods and Force Fields. Journal of Chemical and Theory Computation, 2010. 6(12): p. 3808–3816. [ Links ]
11. Hartshorn, M.J. , et al. , Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem, 2007. 50(4): p. 726-41. [ Links ]
12. Berman, H.M. , et al. , The Protein Data Bank. Nucleic Acids Res, 2000. 28(1): p. 235-42. [ Links ]
13. Morris, G.M. , et al. , Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem, 1998. 19(14): p. 1639-1662. [ Links ]
14. Abreu, R.M. , et al. , MOLA: a bootable, self-configuring system for virtual screening using AutoDock4/Vina on computer clusters. J Cheminform, 2010. 2(1): p. 10. [ Links ]
15. Sanner, M.F. , Python: a programming language for software integration and development. J Mol Graph Model, 1999. 17(1): p. 57-61. [ Links ]
16. Studio, D. , version 2.5. Accelrys Inc. : San Diego, CA, USA, 2009. [ Links ]
17. Steward, J. , MOPAC (2009). Steward computational Chemistry, Version 9.069 W. [ Links ]
