










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Colombiana de Química
versão impressa ISSN 0120-2804
Rev.Colomb.Quim. vol.42 no.3 Bogotá set./dez. 2013
Validación de una metodología analítica empleando espectrofotometría ultravioleta para el estudio de la solubilidad de algunas sulfonamidas en mezclas cosolventes
Validation of an analytical method for the study of the solubility of some sulfonamides in cosolvent mixtures by ultraviolet spectrophotometry
Validação de um método analítico utilizando espectrofotometria de UV para o estudo da solubilidade de algumas sulfamidas em misturas cosolvente + agua
Daniel R. Delgado, Gerson A. Rodríguez, Jorge A. Martínez, Jaime H. Rojas, Fleming Martínez*
Departamento de Farmacia, Facultad de Ciencias, Universidad Nacional de Colombia, Sede Bogotá D.C., A.A. 14490, Bogotá D.C., Colombia *e-mail: fmartinezr@unal.edu.co
Resumen
En este trabajo se propone y se valida una metodología analítica por espectrofotometría UV aplicada al estudio de la solubilidad de algunas sulfonamidas en mezclas cosolventes. Los parámetros evaluados fueron: especificidad, linealidad, precisión y límites de detección y cuantificación, así como la estabilidad de los fármacos bajos las condiciones de análisis de solubilidad. El método propuesto es útil para determinar la solubilidad de estas sulfonamidas en función de la temperatura y la composición cosolvente.
Palabras clave: Validación, espectrofotometría UV-vis, sulfonamidas, solubilidad.
Summary
An analytical method by UV-spectrophotometry has been proposed and validated to study the solubility of some sulfonamides in cosolvent mixtures. The parameters evaluated were specificity, linearity, precision, and detection and quantification limits, as well as the drug stability under the solubility analysis conditions. The developed method was useful to determine the solubility of these drugs as a function of temperature and cosolvent concentration.
Key Words: Validation, UV/VIS-spectrophotometry, sulfonamides, solubility.
Resumo
Neste trabalho propomos é validamos uma metodologia analítica ultravioleta para o estudo da solubilidade de alguns sulfamidas em misturas dos solventes. Os parâmetros avaliados foram: especificidade, linearidade, precisão, exatidão e limites de detecção e quantificação, e a estabilidade dos fármacos nas condições de estúdio. O método proposto é útil para a determinação da solubilidade destes sulfamidas em função da temperatura e da composição do cosolvente.
Palavras-chave: Validação, espectrofotometria UV-vis, sulfamidas, solubilidade.
Introducción
En la actualidad, las sulfonamidas tienen importancia farmacológica relevante en el tratamiento de algunas infecciones causadas por bacterias Gram-positivas y Gram-negativas, algunos hongos y ciertos protozoarios debido a que son inhibidoras competitivas de la enzima dihidropteroato sintetasa bacteriana (1). Esta enzima es necesaria para el procesamiento adecuado del ácido para-aminobenzoico, esencial en la síntesis de ácido fólico. Por esta razón, estas sulfonamidas son consideradas antibióticos de amplio espectro (2, 3). Estudios realizados por Wang et al. exponen la gran importancia de las sulfonamidas en el campo de la química supramolecular y medicinal, mediante el desarrollo de nuevos agentes terapéuticos combinando dos compuestos, entre ellos las sulfonamidas, obteniendo agentes terapéuticos más potentes, de mayor espectro, menos tóxicos, con menores efectos adversos, mejor biodisponibilidad y por tanto mayor seguridad (4). De otro lado, Krátký et al. plantean la posibilidad del diseño de fármacos antituberculosos a partir de varias sulfonamidas, puesto que estas inhiben la β-anhidrasa carbónica a bajas concentraciones en Mycobacterium tuberculosis, lo que propendería a disminuir efectos adversos (5). Lo anterior pone de manifiesto la importancia de los estudios que permitan caracterizar las propiedades fisicoquímicas, dentro de las cuales la solubilidad es una de las más relevantes puesto que es parte esencial en el desarrollo de productos farmacéuticos, ya que afecta sus características biofarmacéuticas, farmacodinámicas y farmacocinéticas (6), por lo que la validación de las metodologías analíticas para la cuantificación de estos fármacos son de gran relevancia para el estudio de su comportamiento químico y biológico (7-9).
Uno de los métodos más utilizados para la determinación de la solubilidad es el denominado método de agitación de frasco, propuesto por Higuchi y Connors (10). Una gran cantidad de valores obtenidos por este método han sido sistematizados recientemente por Jouyban (11). Este método implica el uso de una metodología analítica para la cuantificación del analito de interés, la cual debe ser validada con el ánimo de proporcionar resultados precisos en su aplicación rutinaria (12). En este sentido la ICH, ha concertado los requisitos de validación en dos directrices, la Q2 (R1) y la Q2B (13, 14). La primera resume y define las características necesarias para varios tipos de procedimientos de ensayo, y la segunda describe los datos experimentales requeridos y la interpretación estadística de los mismos. Con base en lo anterior, en este trabajo se realiza la validación de una metodología analítica por espectrofotometría ultravioleta para la cuantificación de sulfadiazina, (SD, 4-amino-N-2-pirimidinilbencensulfonamida, CAS RN: [68-35-9], Fig. 1), sulfamerazina, (SMR, 4-amino-N-(4-metil-2-pirimidinil)-bencensulfonamida, CAS RN: [127-79-7], Fig. 1) y sulfametazina (SMT, 4-amino-N-(3,5-dimetilfenil)-bencensulfonamida, CAS RN [57-68-1], Fig. 1) (15, 16), en cuatro solventes puros, agua (W), metanol (MeOH), etanol (EtOH) y propanol (PrOH), y en mezclas cosolventes (MeOH + W, EtOH + W y PrOH + W), puesto que las tres sulfonamidas presentan grupos cromóforos en su estructura molecular, los cuales permiten obtener una adecuada absorción en la región UV.
Debido a que la técnica de espectrofotometría ultravioleta (UV) no es una metodología específica, ya que no permite discriminar a cuál analito corresponde una lectura, se hace necesario garantizar que en el momento de la determinación de la absorbancia, la muestra corresponda al analito de interés y no a una mezcla de sustancias o productos de degradación; por esta razón se realizó un análisis por cromatografía líquida de alta eficiencia (CLAE) con detector UV/DAD (CLAE-DAD) para verificar la presencia o ausencia de productos que pudieran generarse durante el tiempo de equilibrio de saturación de las sulfonamidas.
Materiales y métodos
Estándares y reactivos
Los estándares sulfadiazina (SD) R.A., sulfamerazina (SMR) R.A, y sulfametazina (SMT) R.A, pureza >99,0% fueron obtenidas de Sigma (E.U.); etanol absoluto R.A Merck (Alemania); metanol Reag. Ph. Eur. HPLC, Merck (Alemania), pureza >99,9%; propanol R.A Merck (Alemania), pureza >99,9%; acetonitrilo Reag. Ph. Eur., Merck (Alemania), pureza > 99,9%; ácido trifluoroacético (TFA) Merck (Alemania), pureza > 99,9%; agua con conductividad menor a 2 mS cm-1, tamiz molecular número 3 y 4 Merck, filtros 0,45 μm Millipore Corp. Swinnex®-13 (E.U.); filtros de membrana de acetato de celulosa 0,3 μm Advantec MFS, Inc. (E.U.); equipo de filtración al vacío; columna Eclipse XDB-C18
Condiciones CLAE
Se empleó un cromatógrafo líquido modular Agilent 1200 Series, con automuestreador Agilent 1260 Infinity, desgasificador Agilent 1200 Series, bomba cuaternaria Agilent 1200 Series, detector UV/VIS con arreglo de diodos e integrador Agilent 1200 Series, así como una columna Eclipse XDB-C18 ( 4,6 mm x 150 mm , 3,5-5 μm) cuya temperatura de operación fue de 25 °C . El volumen de inyección fue de 10 μl; la fase mσvil utilizada fue sistema de gradiente lineal de dos solventes con agua/acetonitrilo, ambos con αcido trifluoroacιtico (TFA) al 0,05% cuya composiciσn de partida fue 90/10 a cero minutos, luego se cambiσ a 85/15 a los diez minutos. La velocidad de flujo fue de 2,5 mL/min, y las longitudes de onda empleadas para la cuantificaciσn fueron de 254 nm y 268 nm, con un ancho de pico de 0,1 minutos, en un rango de escaneo de 190 a 400 nm y un ancho de banda de 1,0 nm con una apertura (slit) de 4 nm.
Determinación de la solubilidad de las sulfonamidas (SD, SMR y SMT)
Para la determinación de la solubilidad termodinámica (equilibrio entre la fase líquida correspondiente a la solución saturada y la fase sólida correspondiente al fármaco no disuelto) de las sulfonamidas, se empleó el método de agitación de frasco propuesto por Higuchi y Connors (10):
Preparación de la muestra: Se adicionó una cantidad suficiente de cada sulfonamida para obtener una solución saturada en equilibrio con la fase sólida a 20 g de solvente (W, MeOH, EtOH y PrOH), contenida en frascos de vidrio ámbar con capacidad de 30 mL y con tapa de polipropileno. Cada muestra, fue sometida a ultrasonido durante 30 minutos antes de ser colocada en el termostato a la temperatura de estudio, posteriormente se agitó periódicamente durante el tiempo de equilibrio.
Separación de fases: Para la separación de fases de la solución saturada, se empleó el método de filtración a través de membranas con diámetro de poro de 0,45 μm para asegurar la ausencia de partículas sólidas, teniendo en cuenta que las jeringas y filtros estuvieran termostatados a la temperatura de estudio. Para reducir los posibles errores en la determinación de la solubilidad por la adsorción del soluto en el filtro, se purgó el mismo con la solución saturada para saturar los sitios de adsorción.
Análisis de la solución saturada: Se realizó un análisis espectrofotométrico UV con las condiciones descritas con anterioridad, para lo cual se tomó una masa de solución de cada una de las muestras, realizando las respectivas diluciones gravimétricas en etanol, luego se determinaron las absorbancias en el espectrofotómetro. Se aseguró, además, que la absorbancia se encontrara en la zona de linealidad de la curva de calibración obtenida para cada sulfonamida.
Validación del método analítico
La validación del método analítico por espectrofotometría UV, se realizó teniendo en cuenta los lineamientos establecidos en la Conferencia Internacional de Armonización (ICH, International Conference on Harmonization, ICH), la farmacopea USP, y las normas de la asociación española de farmacéuticos de la industria (13, 14, 17, 18), analizando los siguientes parámetros:
Especificidad.
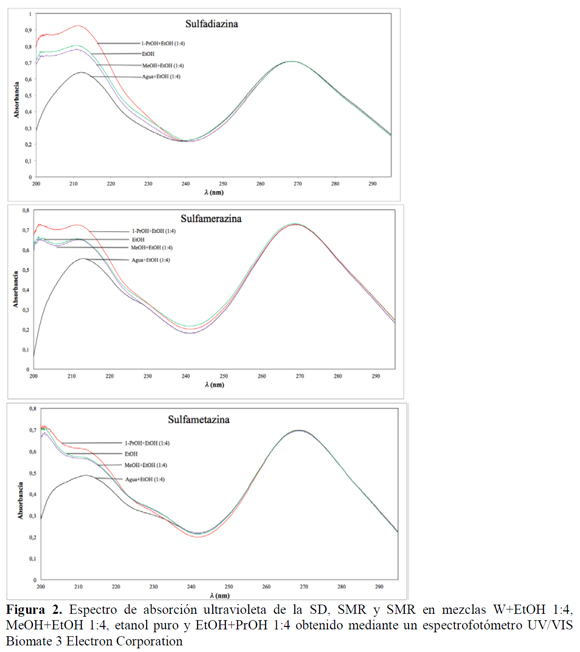
Se validó la capacidad del método analítico para cuantificar cada una de las tres sulfonamidas de forma inequívoca. Inicialmente se determinó el espectro ultravioleta de cada sulfonamida en EtOH, para obtener la longitud de onda de máxima absorbancia (λmax) (Figura 2). De otro lado, si bien es claro que cuando se realiza experimentalmente una determinación con el espectrofotómetro, el mismo solvente de las diluciones es utilizado como blanco, y por lo tanto el equipo descuenta la posible contribución del mismo, en este trabajo se realizó un análisis por CLAE-DAD de los solventes puros empleados en el estudio de solubilidad (MeOH, EtOH y PrOH) a la longitud de onda de máxima absorbancia de los analitos para determinar si éstos absorben en estas condiciones, encontrándose que los cromatogramas obtenidos no muestran ningún pico en esa longitud de onda (Figura 3).
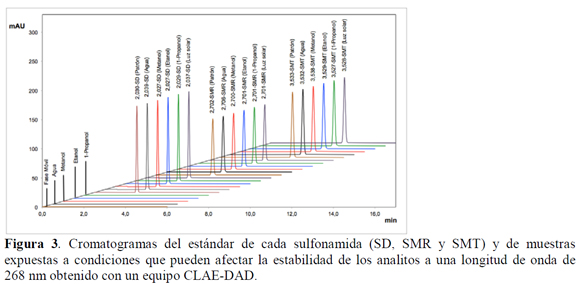
Adicionalmente, con el ánimo de garantizar que al momento de realizar la cuantificación no hubiera presencia de productos de degradación que pudieran interferir en la medida, arrojando datos inexactos, se sometió cada fármaco a condiciones de estrés: exposición a la luz solar directa (solamente en agua) y temperatura de 50 °C en los cuatro solventes empleados en el estudio de solubilidad (W, MeOH, EtOH y PrOH) durante cinco días. Las condiciones de estrés, referidas a la hidrólisis ácida y básica de los analitos, no se realizaron en esta investigación puesto que el objetivo global del presente estudio no consistió en determinar productos de degradación de las sulfonamidas en condiciones extremas de pH, ya que las mismas no se presentan en este tipo de estudios fisicoquímicos. Las muestras fueron analizadas por CLAE-DAD, con el fin de establecer la presencia o ausencia de sustancias de degradación que pudieran interferir de manera directa en las lecturas espectrofotométricas (Figura 3).
El criterio de aceptación de este parámetro es demostrar que la lectura del espectrofotómetro sea consecuencia únicamente de la concentración del fármaco en la dilución correspondiente y no a la presencia de los solventes utilizados o posibles productos de degradación que se puedan generar bajo las condiciones de solubilidad, durante el tiempo de equilibrio de saturación.
Linealidad y rango
Para evaluar la linealidad, se establecieron cinco niveles de concentración (1,5; 4,5; 7,5; 10,5 y 13,5 mg/g para la SD) y (2,0; 5,0; 8,0; 11,0 y 14,0 mg/g para la SMR y la SMT) utilizando etanol absoluto como solvente, realizando tres replicas (niveles de concentración K=5, número de réplicas n=3) para un total de 15 determinaciones por cada sulfonamida. Las muestras se analizaron de manera aleatoria y no de manera creciente para ofrecer un análisis estadístico válido. La evaluación de la linealidad se obtuvo a través del análisis de varianza (ANOVA) para la regresión, y el estadístico t de Student se usó para evaluar la pendiente y el intercepto, para un grado de significancia a de 0,05.
Precisión
La ICH Q2B (14), define como ámbito de aplicación de la precisión, la determinación cuantitativa de principios activos, por lo que se hace necesario evaluar la precisión mediante el estudio de la repetibilidad instrumental y del método, además de la precisión intermedia.
Para evaluar la repetibilidad del equipo, se preparó una solución de SD con una concentración cerca de la concentración nominal (7,5 μg/g) a la cual se le determinó 12 veces su absorbancia. El criterio de aceptación de este parámetro es que el coeficiente de variación de las medidas sea menor al 1,0% (18).
En cuanto a la repetibilidad del método, ésta se analizó a partir de una solución a la concentración nominal de la recta de calibración, realizando 7 réplicas para cada sulfonamida. El criterio de aceptación de este parámetro es que el coeficiente de variación de las medidas sea menor al 1,0% (18).
Finalmente la precisión intermedia, se evaluó considerando dos analistas y realizando las mediciones durante tres días diferentes por triplicado, lo que implica la preparación de cada muestra desde la pesada. El criterio de aceptación para este parámetro es que el coeficiente de variación de las medidas sea menor que el doble del coeficiente de variación correspondiente a la repetibilidad del método (18).
Los resultados del estudio de linealidad se tabularon relacionando las concentraciones y su respuesta instrumental (absorbancia), expresando su relación matemáticamente como una recta de regresión lineal de la forma y = a + bx, obtenida mediante el método de ajuste de mínimos cuadrados.
Límite de detección (LD) y límite de cuantificación (LC)
Los límites de detección y cuantificación se determinaron a partir de la pendiente y la desviación estándar del intercepto de la curvas de calibración en el rango de linealidad para cada una de las sulfonamidas (14, 18, 19-21).
Estabilidad
Con el fin de determinar la estabilidad de las tres sulfonamidas en aquellas condiciones a las que estarían expuestas durante el ensayo de solubilidad, se prepararon un total de 15 muestras (tres de cada sulfonamida en cada alcohol, y seis en agua, de las cuales tres se expusieron a luz solar directa, garantizando el equilibrio termodinámico de saturación (presencia de dos fases sólido-líquido) y se termostatizaron las muestras a 50 °C durante 120 h (5 días). Las áreas obtenidas por CLAE-DAD de SD, SMR y SMT determinadas al comienzo de cada ensayo se compararon con las obtenidas al final del ensayo con el fin de identificar su posible degradación.
Resultados y discusión
Las medidas espectrofotométricas se realizaron en etanol absoluto, puesto que la solubilidad de las tres sulfonamidas (SD, SMR y SMT) en este solvente es alta, reduciendo el riesgo de precipitación; por esta razón las diluciones de cada una de las muestras se hicieron en este alcohol, a pesar de que la matriz inicial puede ser un solvente o mezcla de solventes diferentes de etanol. Se evaluó, además, la influencia de todos los solventes empleados en la respuesta analítica, como los efectos batocrómico, hipsocrómico, hipercrómico o hipocrómico además de verificar si estos solventes absorbían o no a la longitud de onda de máxima absorbancia de cada fármaco (Figura 2).
Para determinar la longitud de onda de máxima absorbancia, se realizó un barrido en la región ultravioleta, en el rango de 200 a 300 nm, definiéndose que el λmax para las sulfonamidas en estudio es de 268 nm (Figura 2). De otro lado, para evaluar el efecto de la presencia de solventes diferentes al etanol (W, MeOH y PrOH), sobre la longitud de onda de máxima absorción, se prepararon tres soluciones de cada sulfonamida a una concentración aproximada de 10 μg/mL en mezclas cosolventes con una fracción másica de 0,20 de agua, metanol o propanol en etanol, en donde se observa un punto isosbéstico a 268 nm (λmax) (Figura 2), demostrándose que los solventes en una relación 1:4 no afectan la cuantificación de las sulfonamidas a la longitud de onda de estudio, teniendo presente que la mínima dilución se realiza para la cuantificación de la SD en agua en una relación 1:4 de etanol y agua.
Adicionalmente, se evaluó si los solventes empleados en el estudio de solubilidad absorbían a la longitud de onda de máxima absorbancia mediante cromatografía de líquidos de alta eficiencia, demostrándose que su incidencia en la cuantificación de las tres sulfonamidas es despreciable (Figura 3).
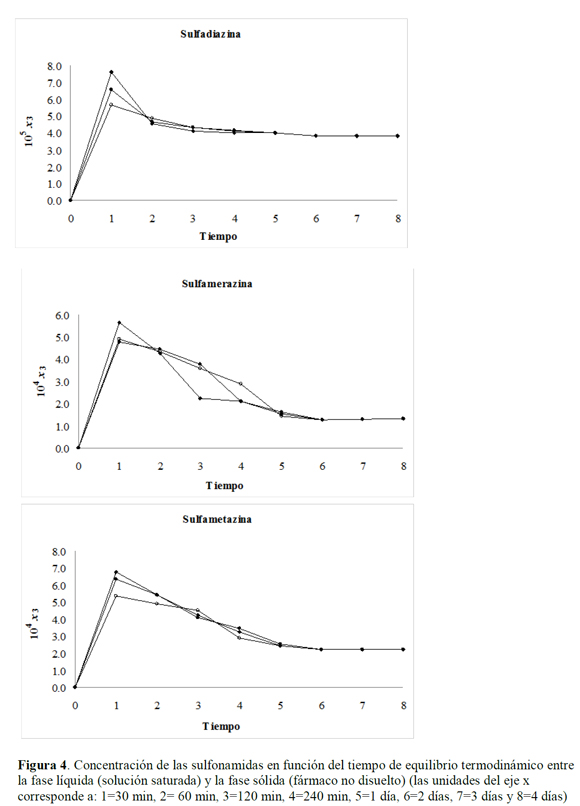
En el estudio de solubilidad de las sulfonamidas se evaluaron cuatro factores que pueden afectar la estabilidad de los fármacos, esto es, la luz solar (los estudios de solubilidad se llevan a cabo en frascos ámbar para reducir el efecto de la luz), la temperatura, los solventes y el tiempo de equilibrio termodinámico el cual se estableció en 72 horas (momento en el cual se realiza la cuantificación de cada una de las sulfonamidas), de acuerdo a la Figura 4, en la cual se evidencia un aumento significativo en la concentración de las soluciones después de ser sometidas a ultrasonido durante los 30 minutos; este aumento vertiginoso en la concentración es posiblemente debido a la reducción en el tamaño de partícula y, por ende, en el aumento del área superficial, lo que implica un aumento en la velocidad de disolución; además, se presenta un aumento en la temperatura del sistema por lo que la concentración determinada a los 30 minutos corresponde a un solubilidad transitoria o temporal, la cual es mayor que la solubilidad termodinámica del fármaco en las condiciones de equilibrio (25 °C) (22-24).
Puesto que la mayor temperatura de los estudios de solubilidad es 40 ºC, y el tiempo de equilibrio termodinámico es de 3 días, para la presente investigación las muestras fueron termostatadas durante 5 días a 50 ºC, con el ánimo de garantizar que en condiciones más drásticas a las del estudio de solubilidad las sulfonamidas no presenten degradación. Inicialmente, se determina que el tiempo de retención para cada sulfonamida es, SD: 2,0 min, SMR: 2,7 min y SMT: 3,5 min (Figura 5), y posteriormente se realizaron cromatografías de las muestras, determinándose las áreas correspondientes a cada fármaco al final del ensayo y fueron comparadas con el área del estándar de cada fármaco control (SD, SMR y SMT) (Figura 3).
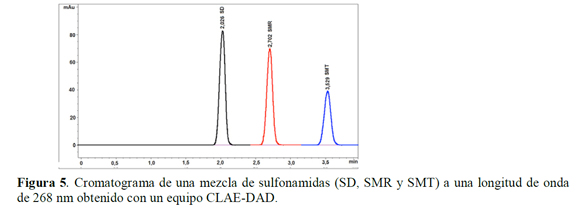
De acuerdo a la tabla 1 se observa que no hay una variación significativa en las áreas de cada uno de los picos de los analitos (porcentajes de coeficientes de variación para la SD, SMR y SMT: 1,44, 0,86, y 1,08% respectivamente) con respecto al estándar de cada sulfonamida; además, los tiempos de retención son muy similares, los cuales presentan porcentajes de coeficiente de variación menores al 0,15% y el factor de pureza de pico indica una alta probabilidad de que la señal corresponda al analito respectivo (> 999,81±0,05), demostrándose así que las tres sulfonamidas son estables en las condiciones del estudio de solubilidad puesto que no se evidencia degradación de las mismas a 50 ºC (temperatura 10 ºC superior a la máxima temperatura de estudios de solubilidad) durante 5 días (tiempo de equilibrio termodinámico: 3 días).
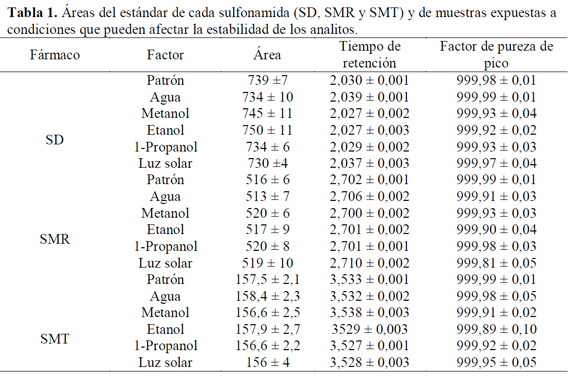
De acuerdo a lo anterior, se demuestra que existe una alta probabilidad de que en el momento de hacer la determinación de la solubilidad de las tres sulfonamidas a cualquier temperatura de estudio de solubilidad (20, 25, 30, 35 y 40 ºC) y en cualquier mezcla cosolvente (MeOH + W, EtOH + W y PrOH + W) a los tres días de equilibrio termodinámico, la lectura de la absorbancia sea consecuencia únicamente de la concentración de cada uno de los analitos y no a productos de degradación de los mismos ni a la presencia de los solventes diferentes al etanol.
Las curvas de calibración se realizaron en etanol (Figura 6), para cada una de las sulfonamidas con el fin de establecer la regresión entre la relación de la concentración de las soluciones de cada sulfonamida (eje "x" expresado en µg/g) y su respuesta instrumental (eje "y" expresado en unidades de absorbancia).
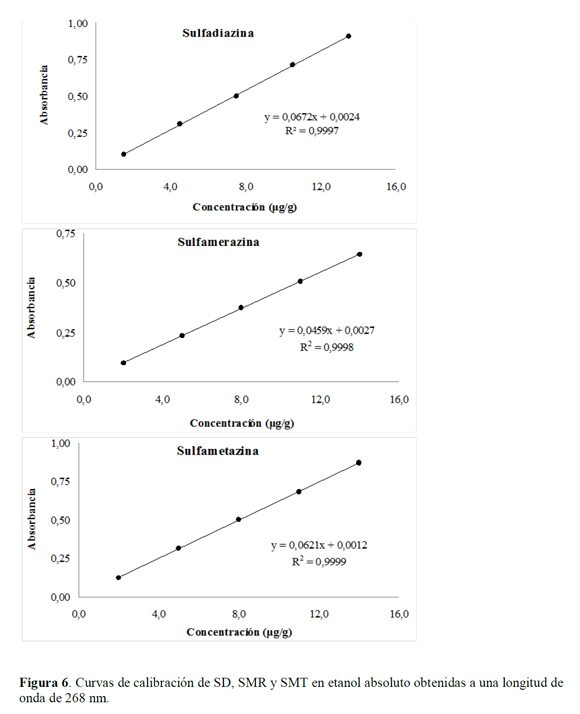
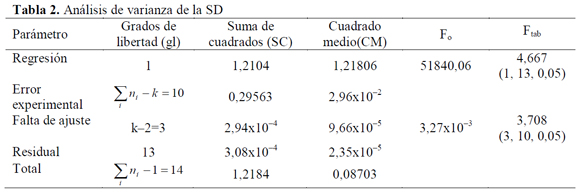
Los resultados del análisis de la linealidad se muestran en las Tablas 2 a 4. Mediante un análisis de varianza (ANOVA), la homocedasticidad se demuestra aplicando la prueba G del test de Cochran, donde Gexp (SD=0,44; SMR=0,45 y SMT=0,15) es menor al Gtab (a=0,05, K=5, n=3)=0,68, lo que significa que las varianzas para los diferentes niveles de concentración son homogéneas, mostrando así que el nivel de concentración no influye en la variabilidad de los resultados para los rangos de concentración definidos, por lo tanto no se presenta una relación estadísticamente significativa entre la variable explicativa (concentración) y el error experimental. En cuanto al análisis de varianza para la regresión, mediante el test F, el Fexp es mayor a Ftab=4,667 con 1 grado de libertad en el numerador correspondiente a la regresión y con 13 grados de libertad en el denominador correspondientes al error residual, indicando que la pendiente es significativamente distinta de cero; y para la falta de ajuste debido a que Fexp es menor a Ftab=3,708 con 3 grados de libertad en el numerador y con 10 grados de libertad en el denominador correspondientes al error experimental, se demuestra linealidad entre los resultados obtenidos de la relación entre la concentración y la respuesta instrumental (absorbancia), para cada sulfonamida con un nivel de significación α=0,05.
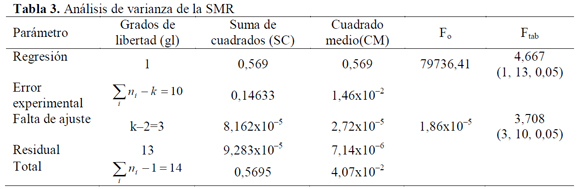
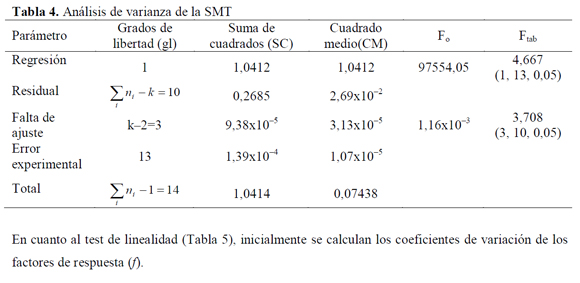
En cuanto al test de linealidad (Tabla 5), inicialmente se calculan los coeficientes de variación de los factores de respuesta (f).
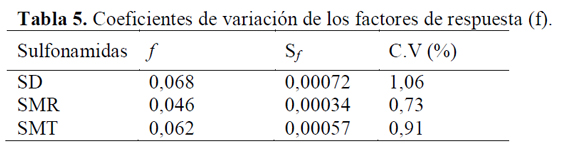
En todos los casos, los promedios de f son aproximados a las pendientes, y los coeficientes de variación son menores al 2%, lo que indica que la calibración es lineal. En cuanto a la prueba t de Student para la pendiente, se confirma lo establecido con el análisis de varianza, además la pendiente es significativamente diferente de cero para todas las sulfonamidas para un grado de significación α=0,05 (Tablas 6 a 8) puesto que texp > ttab =2,16 para la pendiente.



El test de proporcionalidad, indica que el intercepto no es significativamente diferente de cero puesto que texp < ttab =2,16 para el intercepto. Finalmente los coeficientes de correlación para la SD, SMR y SMT son 0,9999, 0,9998 y 0,9999 respectivamente; de otro lado, texp > ttab =2,16 para el coeficiente de correlación, indicando que éste es significativamente diferente de cero, mostrando que los datos experimentales se ajustan al modelo de regresión lineal. De acuerdo a las pruebas t de Student, se rechaza la hipótesis nula para pendiente y parámetro de correlación y se acepta para el intercepto.
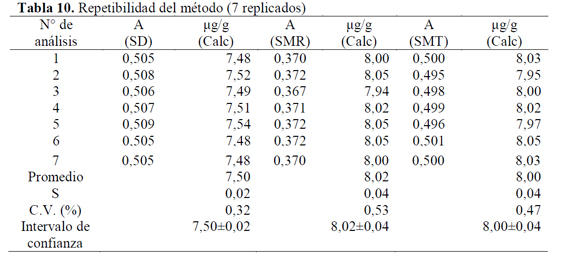
Para evaluar la precisión del método se evaluó la repetibilidad del instrumento y del método mismo, obteniendo para el primer caso, que el coeficiente de variación de las 12 lecturas de una solución de SD de 7,512 μg/g en etanol, fue de 0,170 %, el cual es menor al porcentaje establecido como máximo en la literatura, el cual es, generalmente del 1,0% (Tabla 9) (25).
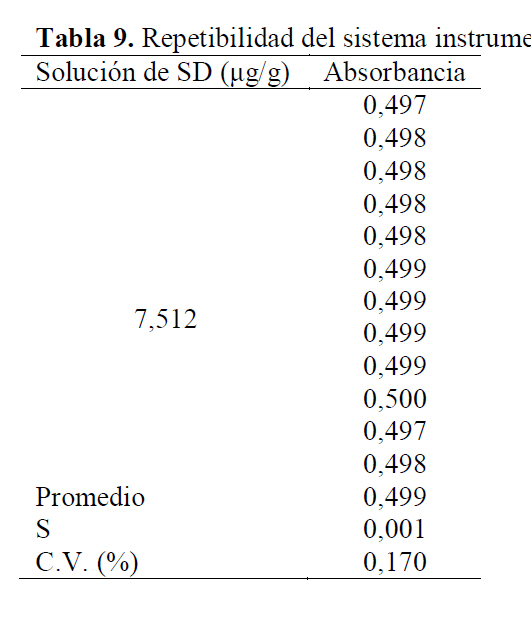
Con respecto a la repetibilidad del método, el coeficiente de variación para las 7 soluciones de SD, SMR y SMT en etanol fueron de 0,32%, 0,53% y 0,47% respectivamente, los cuales se encuentran dentro de los intervalos máximos permitidos y que oscilan entre el 2,0 y el 3,0% (Tabla 10) (26). De acuerdo con estos resultados se puede afirmar que la metodología cumple con el parámetro de repetibilidad. Ahora bien, la ICH Q2B recomienda introducir los intervalos de confianza en los estudios de precisión (14), calculados como X ± tS/√n , en donde X es la media de la serie de resultados obtenidos en un mismo nivel de concentración, t es el valor de la t Student de las tablas para n-1 grados de libertad y a=0,05, n es el número de análisis y s es la desviación estándar (18).
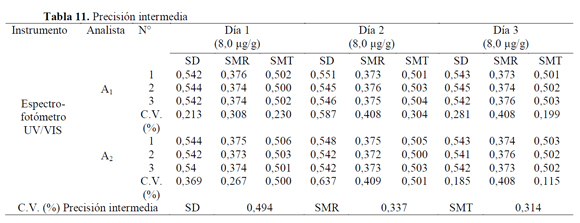
Para el análisis de la precisión intermedia se evaluó una concentración de 8,00 mg/g para cada una de las sulfonamidas, para lo cual se desarrolló un diseño experimental variando los factores analista y día del análisis (Tabla 11). Los coeficientes de variación globales de las respuestas obtenidas para SD, SMR y SMT fueron 0,494%, 0,337% y 0,314%, respectivamente. La prueba de Cochran, demuestra que los factores: analista y día del ensayo, no contribuyen a que se presente una diferencia estadísticamente significativa entre varianzas de las respuestas (absorbancias) de los ensayos, puesto que Gexp (SD=0,043; SMR=0,200 y SMT=0,057) es menor al Gtab (a=0,05, K=6, n=3)=0,6161 indicando homogeneidad de varianzas.
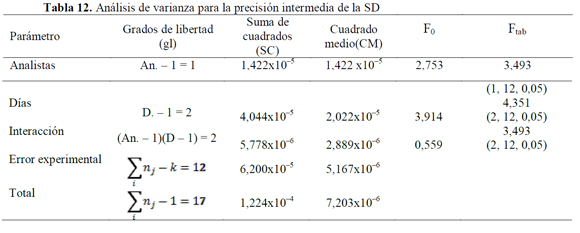
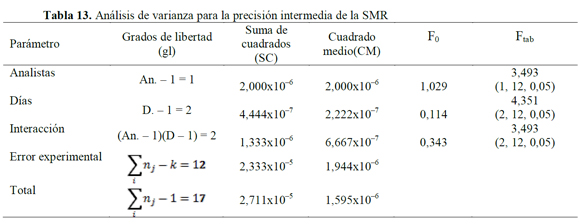
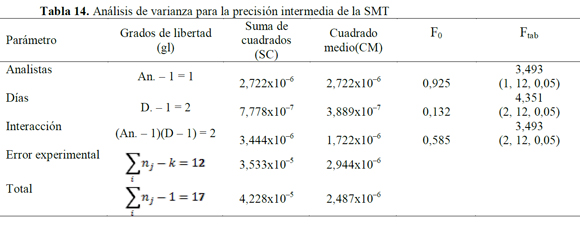
El análisis de varianza (ANOVA), usando un modelo de efectos o categorías fijas para el cálculo de F (27), mostró que valores de Fexp son mayores a los Ftab (Tablas 12, 13 y 14), indicando que los resultados no presentan una diferencia estadísticamente significativa, cuando el ensayo es realizado por un analista en particular, o cuando se varía el día en que este se realiza (28, 29); además, la variabilidad de los resultados no se debe a determinadas combinaciones día de análisis-analista.
Conclusiones
Con base en los parámetros analíticos evaluados, el método espectrofotométrico en el ultravioleta propuesto es adecuado para la cuantificación de las tres sulfonamidas estudiadas (SD, SMR y SMT), en estudios de solubilidad en las mezclas cosolventes MeOH + W, EtOH + W y PrOH + W, y además, en los solventes puros, mediante el método de agitación de frasco. De hecho, el método propuesto ya ha sido utilizado en la determinación de la solubilidad de estas tres sulfonamidas en mezclas cosolventes etanol + agua a diferentes temperaturas (30, 31).
Agradecimientos
Al Departamento de Farmacia de la Universidad Nacional de Colombia por facilitar los equipos e instalaciones utilizados en el desarrollo de esta investigación.
Bibliografía
1. Delgado, A.; Minguillón, C.; Joglar, J. Introducción a la Química Terapéutica. 2a ed. Madrid: Diaz de Santos S.A., 2003, p. 461-467. [ Links ]
2. Perlovich, G.L.; Ryzhakov, A.M.; Strakhova, N.N.; Kazachenko, V.P.; Schaper, K.J.; Raevskya, O.A. Thermodynamic aspects of solubility and partitioning processes of some sulfonamides in the solvents modeling biological media. J. Chem. Thermodynamics. 2014. 69: 56-65. [ Links ]
3. Delgado, D.R. Estudio termodinámico de la solubilidad de algunas sulfonamidas sódicas en mezclas cosolventes etanol + agua. Tesis de Maestría. Universidad Nacional de Colombia, Bogotá D.C., 2010. [ Links ]
4. Wang, X.L.; Wan, K.; Zhou, C.H. Synthesis of novel sulfanilamide-derived 1,2,3-triazoles and their evaluation for antibacterial and antifungal activities. Eur. J. Med. Chem. 2010. 45: 4631-4639. [ Links ]
5. Krátký, M.; Vinsová, J.; Volková, M.; Buchta, V.; Trejtnar, F.; Stolaríková, J. Antimicrobial activity of sulfonamides containing 5-chloro-2-hydroxybenzaldehyde and 5-chloro-2-hydroxybenzoic acid scaffold. Eur. J. Med. Chem. 2012. 50: 433-440. [ Links ]
6. Aulton, M.E. Farmacia: La Ciencia del Diseño de las Formas Farmacéuticas. 2a ed. Madrid: Elsevier, 2004. [ Links ]
7. Yu, H.; Tao, Y.; Chen, D.; Wang, Y.; Huang, L.; Peng, D., Dai, M.; Liu, Z.; Wang, X.; Yuan, Z. Development of a high performance liquid chromatography method and a liquid chromatography-tandem mass spectrometry method with the pressurized liquid extraction for the quantification and confirmation of sulfonamides in the foods of animal origin. J. Chromatogr. B 2011. 879: 2653-2662. [ Links ]
8. Yun-Hong, H.; Yang, X.; Qing-Hua, H.; Yu-Sheng, C.; Bi Bai, D. Determination of sulfadiazine residues in pork by molecular imprinted column coupling with high performance liquid chromatography. Chin. J. Anal. Chem. 2012. 40: 1011-1018. [ Links ]
9. Cioroiu, B.I.; Lazar, M.; Bello-López, M.A.; Fernández-Torres, R. Identification of the specified impurities of silver sulfadiazine using a screening of degradation products in different stress physico-chemical media. Talanta. 2013. 116: 653-662. [ Links ]
10. Higuchi, T., Connors, K.A. Phase-solubility techniques. En:Advances in Analytical Chemistry and Instrumentation, vol. 4. New York: John Wiley & Sons, 1965, pp. 117-212. [ Links ]
11. Jouyban, A. Handbook of Solubility Data for Pharmaceuticals. New York: CRC Press, Taylor & Francis Group, 2010. [ Links ]
12. Ermer, J. Validation in pharmaceutical analysis. Part I: An integrated approach, J. Pharm. Biomed. Anal. 2001. 24: 755-767. [ Links ]
13. ICH Q2 (R1): Validation of Analytical Procedures, Text and methodology, November 2005. [ Links ]
14. ICH Q2B: Analytical Validation-Methodology, November 1996. [ Links ]
15. Budavari, S.; O'Neil, M.J.; Smith, A.; Heckelman, P.E.; Obenchain Jr., J.R.; Gallipeau, J.A.R.; D'Arecea, M.A. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals. 13th edition. Whitehouse Station, NJ: Merck & Co., Inc., 2001, p. 1586-1589. [ Links ]
16. Moffat, A.C.; Jackson, J.V.; Moss, M.S.; Widdop, B. Clarke´s Isolation and Identification of Drugs: In Pharmaceuticals, Body Fluid, and Post-Mortem Material. London: The Pharmaceutical Press, 1986, p. 979. [ Links ]
17. US Pharmacopeia.30th revision. Rockville, MD: United States Pharmacopeial Convention, Inc., 2007, p. 680. [ Links ]
18. Aguirre, L.; Garcia, F.J.; Junca, T., Fontanet, M.; Roca, M.; Carbó, M.; Sanfeliu, A.; Pomar, M.; Pérez, P.; Ochoa, C.; Sánchez, M.; Forn, M.; Serrat, M.; Gonfaus, M. Validación de Métodos Analíticos. Barcelona: AEFI, 2001. [ Links ]
19. Vessman, J. Selectivity or specificity? Validation of analytical methods from the perspective of an analytical chemist in the pharmaceutical industry. J. Pharm. Biomed. Anal. 1996. 14: 867-869. [ Links ]
20. Persson, B.A.; Vessman, J. The use of selectivity in analytical chemistry - Some considerations. Trends Anal. Chem. 2001. 20: 526-532. [ Links ]
21. Gómez, S.M.; Martínez, J.A.; Martínez, F. Validación de un método analítico empleando cromatografía líquida de alta eficiencia para la determinación de ibuprofeno en medios biorrelevantes. Quim. Nova 2010. 33: 1794-1799. [ Links ]
22. Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 1897. 19: 930-934. [ Links ]
23. Dokoumetzidis, A.; Macheras, P. A century of dissolution research: From Noyes and Whitney to the Biopharmaceutics Classification System. Int. J. Pharm. 2006. 321: 1-11. [ Links ]
24. Siepmann, J.; Siepmann, F. Mathematical modeling of drug dissolution. Int. J. Pharm. 2013. 453: 12-24. [ Links ]
25. Mora, C.P.; Tello, M.E.; Martínez, F. Validación de una metodología analítica para la cuantificación de naproxeno en estudios de reparto líquido/líquido mediante espectrofotometría ultravioleta. Rev. Colomb. Cienc. Quím. Farm. 2006. 35: 81-105. [ Links ]
26. Association of Official Analytical Chemists. Statistical Manual of the AOAC, 12th ed. Washington, 1975, p. 1094. [ Links ]
27. Morales, P. Análisis de variancia con dos criterios de clasificación (diseños factoriales). Facultad de Ciencias Humanas y Sociales, Universidad Pontificia Comillas, España, 2009. [ Links ]
28. Morantes, M.; Vargas, M.; Figueroa, F.S., Sierra, N.; Barbosa, H. Desarrollo y validación de una metodología analítica por HPLC para la cuantificación de fenobarbital en una suspensión extemporánea. Rev. Colomb. Cienc. Quím. Farm. 2010. 39: 68-78. [ Links ]
29. Berrio, M.; Trujillo, M.; Vallejo, B.M.; Barbosa, H.J. Desarrollo y validación de una metodología analítica por HPLC-DAD para la cuantificación de warfarina sódica en una preparación extemporánea. Rev. Colomb. Cienc. Quím. Farm. 2013. 42: 122-133. [ Links ]
30. Delgado, D.R.; Martinez, F. Solution thermodynamics of sulfadiazine in ethanol + water mixtures. J. Mol. Liq. 2013. 187: 99-105. [ Links ]
31. Delgado, D.R.; Martínez, F. Solubility and solution thermodynamics of sulfamerazine and sulfamethazine in some ethanol + water mixtures. Fluid Phase Equilib. 2013. 360: 88-96. [ Links ]
