










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Colombiana de Química
versão impressa ISSN 0120-2804
Rev.Colomb.Quim. vol.43 no.1 Bogotá jan./jun. 2014
https://doi.org/10.15446/rev.colomb.quim.v43n1.50538
DOI: http://dx.doi.org/10.15446/rev.colomb.quim.v43n1.50538
Comparación de dos metodologías para la determinación de residuos de plaguicidas en agua potable
Comparison of two methodologies for the determination of pesticides residues in drinking water
Comparação de metodologias para a determinação de resíduos de pesticidas em água potável
Jairo Arturo Guerrero Dallos1, Nancy Yohanna Velandia Rodriguez1
1Laboratorio de Análisis de Residuos de Plaguicidas - LARP, Departamento de Química, Facultad de Ciencias, Universidad Nacional de Colombia. A. A. 14490, Bogotá, Colombia.
Correo electrónico de contacto: jaguerrerod@unal.edu.co
Recibido: 17 noviembre 2013. Aceptado: 20 enero 2014
Resumen
Se realizó un estudio comparativo de dos metodologías para la determinación de residuos de plaguicidas en agua potable con el fin de establecer cuál de ellas es más adecuada como metodología de análisis. Las metodologías evaluadas involucraron extracción en fase sólida empleando el adsorbente polimérico LiChrolut®EN y extracción líquido-líquido utilizando n-hexano como solvente de extracción. La determinación se realizó por cromatografía de gases con detectores µ-ECD y NPD. El estudio involucró 50 plaguicidas, entre los que se encuentran diferentes familias químicas, tales como organofosforados, organoclorados y piretroides entre otros. Para comparar las metodologías, se evaluó la capacidad de detección y cuantificación estableciendo los límites de detección y cuantificación; la exactitud con experimentos de recuperación y la dispersión por medio de la precisión. Se estableció que estas presentan detección adecuada frente a las concentraciones máximas permitidas por la normativa colombiana, excepto para el insecticida monocrotofos, para el cual se obtuvo un límite de detección superior al exigido. Los experimentos de recuperación mostraron que la totalidad de plaguicidas organoclorados y piretroides pueden ser analizados empleando extracción líquido-líquido, mientras que la extracción en fase sólida es más adecuada para los plaguicidas más polares, tales como los organofosforados y azoles. Se determinó que 22 compuestos se pueden analizar por cualquiera de las dos metodologías, que por extracción en fase solida es posible analizar 36 de los 50 compuestos evaluados mientras que empleando extracción líquido-líquido se pueden analizar 28 de ellos.
Palabras clave: Plaguicidas, agua potable, extracción líquido-líquido, extracción en fase sólida.
Abstract
A comparative study of two methodologies for the determination of pesticide residues in drinking water was performed in order to establish which of them is more suitable of a method of analysis. The assessed methodologies involved solid phase extraction using polymeric adsorbent LiChrolut ® EN and liquid-liquid extraction using n-hexane as the extraction solvent; its determination was performed by GC with µECD and NPD detectors. The study involved 50 pesticides, including different chemical families, such as organophosphates, organochlorines and pyrethroids among others. The detection capability was assessed by establishing the limits of detection and quantification, and the accuracy and precision performing recovery experiments. It was determined that the two methodologies have suitable detection regarding the maximum concentrations permitted by the Colombian regulations, except for monocrotophos insecticide, for which a limit of detection obtained was slightly above to the required value. The recovery experiments showed that all organochlorine and pyrethroid pesticides can be analyzed using liquid-liquid extraction, while the solid phase extraction is more suitable for the more polar pesticides such as organophosphates and azoles; It was determined that 22 compounds can be analyzed by either of the two methods, that is possible to analyze 36 of the 50 compounds evaluated by solid phase extraction, while using liquid liquid extraction 28 can be analyzed.
Keywords: Pesticides, drinking water, liquid-liquid extraction, solid phase extraction.
Resumo
Um estudo comparativo de dois métodos foi realizada para a determinação de resíduos de pesticidas em água potável, a fim de estabelecer qual deles é mais adequado como um método de análise. As metodologias avaliadas envolvendo extracção em fase sólida, utilizando o adsorvente polimérico LiChrolut®EN e extracção líquido-líquido utilizando n-hexano como solvente de extracção. A determinação foi realizada por detectores de GC-ECD e NPD. O estudo envolveu 50 pesticidas, incluindo aqueles que são diferentes famílias químicas, tais como os organofosforados, organoclorados e piretróides, entre outros. Para comparar os métodos, a capacidade de detectar e quantificar a estabelecer os limites de detecção e quantificação foi avaliada; a precisão e as experiências de recuperação usando precisão dispersão. Foi estabelecido que estes apresentaram taxas de detecção adequados para as concentrações máximas permitidas pela legislação colombiana, com exceção de monocrotofós inseticida, para que requerido foi obtido um limite de detecção de. Ensaios de recuperação, mostraram que todos os pesticidas organoclorados e piretróides podem ser analisados utilizando-se extracção líquido-líquido, enquanto que a extracção da fase sólida é mais adequado para os pesticidas mais polares, tais como organofosfatos e azóis. Determinou-se que 22 compostos podem ser analisados por qualquer um de dois métodos, que a extracção de fase sólida, é possível analisar 36 dos 50 compostos avaliados, enquanto utilizando a extracção liquido-liquido 28 pode analisar.
Palavras-Chave: Pesticidas, água potável, extração líquido-líquido, extração em fase sólida.
Introducción
El acceso a fuentes de agua de calidad tiene una incidencia directa sobre la salud de la población. En Colombia la calidad del agua potable está regulada por la resolución 2115 de junio de 2007 del Ministerio de la Protección Social e indica en su artículo 8 las concentraciones máximas de plaguicidas y otras sustancias tóxicas en el agua potable, establecidas según su toxicidad y peligrosidad. Para garantizar que el agua potable cumple con dichas características de inocuidad es necesario contar con medidas de control en campo las cuales se verifican con metodologías confiables que permitan la determinación de contaminantes a los niveles establecidos en la reglamentación, como es el caso de los plaguicidas, que son ampliamente usados en la agricultura y que frecuentemente, por su uso incorrecto pueden ir a contaminar fuentes hídricas tanto superficiales como subterráneas (1).
Existe una gran variedad de métodos para la determinación de plaguicidas en aguas, algunos de ellos desarrollados con el fin de determinar grupos de compuestos en particular, como es el caso de los plaguicidas organoclorados, que tienen gran importancia desde el punto de vista medioambiental debido a su elevada persistencia, o los organofosforados que debido a su elevada toxicidad aguda en mamíferos son considerados relevantes desde el punto de vista toxicológico (2).
Una de las metodologías más comunes es la extracción líquido - líquido, en la cual se utilizaron inicialmente solventes de baja polaridad tales como el diclorometano, el hexano y el éter de petróleo para la extracción de plaguicidas organoclorados y organofosforados (1, 3). En 2010 Wu et al. (4), desarrollaron una metodología para el análisis de piretroides mediante extracción líquido – líquido, empleando 60 mL de diclorometano como solvente de extracción. Para la determinación de plaguicidas organoclorados principalmente se valen del uso de n-hexano (5), éter etílico o cloruro de metileno (6) utilizando entre 25 y 300 mL. Las metodologías desarrolladas eran adecuadas para el propósito, sin embargo tenían como desventaja el uso de grandes volúmenes de solvente, generando cantidades de residuos contaminantes, además de ser dispendiosas en su ejecución experimental debido a la formación de emulsiones durante el análisis. Posteriormente se desarrollaron metodologías analíticas más amigables con el ambiente en las que se redujo considerablemente el uso de solventes de elevada toxicidad y se disminuyó significativamente el tiempo de análisis, incrementando a la vez el número de analitos del alcance de las metodologías (6).
Actualmente la tendencia en el análisis de plaguicidas en aguas involucra el uso de solventes de menor toxicidad y con cantidades pequeñas de estos, tales como la extracción líquido – líquido con n-hexano (7) y la extracción líquido – líquido miniaturizada en la que se usan tan solo unos cuantos microlitros de solvente para realizar la extracción (8). Ésta metodología ha sido utilizada para la determinación de plaguicidas organofosforados en aguas empleando como solvente de extracción una mezcla de acetonitrilo y 1-dodecanol, obteniendo porcentajes de recuperación elevados (90-99%) y bajos límites de detección (5 x 10-4 – 5 x 10-1 mg/L), empleando para la separación y detección cromatografía líquida con detección DAD (9). Así mismo, la microextracción líquido – líquido asistida con aire empleando tolueno como solvente de extracción, fue utilizada por Farajzadeh et al. (10), para analizar plaguicidas de la familia de los triazoles en aguas superficiales y matrices vegetales empleando cromatografía de gases y detección de ionización de llama, obteniendo bajos límites de detección y cuantificación. Para la determinación de herbicidas del grupo de los fenoxiácidos, Jimenez (11) empleó pequeños volúmenes de muestra (10 mL) y acetona como solvente de extracción (3,5 mL) para realizar una extracción líquido – líquido y posteriormente una limpieza en fase sólida dispersiva con octadecilsilano para eliminar interferencias, obteniendo porcentajes de recuperación entre el 82% y el 114%.
Como técnica alterna, la extracción en fase sólida se popularizó para el análisis de muestras ambientales debido a sus grandes ventajas en cuanto a la disminución del uso de solventes, la rapidez de su ejecución y su capacidad para concentrar los analitos, lo cual permitió la determinación de menores concentraciones (2, 6, 12). En el ámbito instrumental se ha utilizado la cromatografía de gases para la determinación principalmente, de plaguicidas volátiles y térmicamente estables como son los plaguicidas organoclorados, organofosforados y piretroides. Los detectores más usados han sido el detector de captura electrónica, actualmente detector de micro captura electrónica (µ-ECD), detectores como el detector de nitrógeno fósforo y el fotométrico de llama, los cuales son más selectivos; actualmente el detector selectivo de masas) es el más utilizado, (5, 13) incluso empleando
metodologías de extracción en fase sólida “on line” acopladas al instrumento, lo que permite minimizar el tiempo de preparación de la muestra, la cantidad de muestra y el consumo de reactivos y solventes (14).
Actualmente la cromatografía de gases acoplada a espectrometría de masas en tandem con analizador de triple cuadrupolo (11) o cuadrupolo acoplado a trampa de iones (15), permite analizar un gran número de compuestos, con mayor selectividad y a concentraciones más bajas (16). Para compuestos con características fisicoquímicas de solubilidad, volatilidad y peso molecular que no les permiten ser incluidos en las metodologías multiresiduo, como es el caso del herbicida glifosato y su metabolito, AMPA, se han desarrollado metodologías específicas empleando extracción en fase sólida y posterior análisis en cromatografía líquida con derivatización post columna y detección con fluorescencia, obteniendo porcentajes de recuperación entre 70% y 73% y límites de detección de 4 x 10-3 mg.L-1 (17).
Con el propósito de contar con metodologías rápidas y adecuadas para análisis de residuos de plaguicidas en agua potable, en este estudio se realizó una comparación de dos metodologías de extracción para la determinación de residuos de 50 plaguicidas de diferentes familias químicas, entre los que se encuentran insecticidas y fungicidas ampliamente utilizados en Colombia.
Materiales y métodos
Estándares, reactivos y soluciones
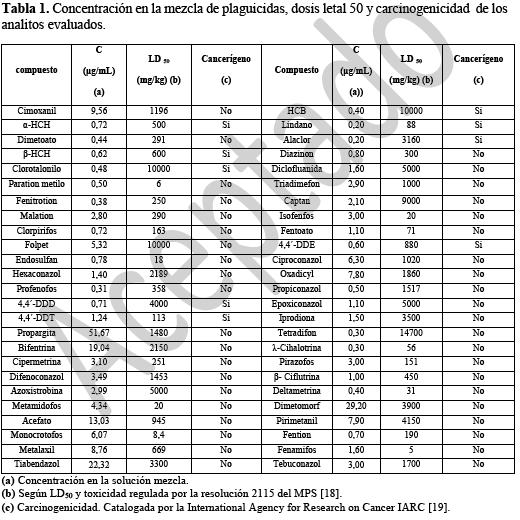
Se emplearon estándares de plaguicidas proporcionados por el Dr. Ehrenstorfer y por Chemservice con una pureza mayor al 95%; Las soluciones madre fueron preparadas en acetato de etilo a concentraciones aproximadas de 500 μg mL-1 y almacenadas en frascos ámbar a -20°C. A partir de las soluciones madre se prepararon mezclas de plaguicidas para los diferentes ensayos. Los analitos evaluados, sus concentraciones y características toxicológicas relevantes se muestran en la tabla 1.
Se emplearon solventes y reactivos marca J.T. Baker grado residuos. Para la extracción en fase sólida se emplearon cartuchos LiChrolut®EN marca Merck de 200 mg de fase estacionaria y 3 mL de capacidad empacados en tubos de polipropileno.
Instrumentos y equipos
Para la separación, identificación y cuantificación de los compuestos se utilizó un cromatógrafo de gases HP6890+ con inyector automático 7683 marca Agilent Technologies® (Palo Alto, CA, EUA). Se utilizó un inyector Split/splitless conectado por medio de una columna capilar sin fase estacionaria (1,0 m, 0,25 mm id), a un divisor de flujo de vidrio silanizado en forma de “Y” unido a una columna HP- 5 (30 m, 0,25 mm id, 0,25 µm df) conectada a un detector µ-ECD 63Ni y una columna HP-50 (30 m, 0,25 mm id, 0,25 µm) conectada un detector NPD. Se utilizó nitrógeno UAP como gas de transporte. Se realizó la inyección en modalidad splitless pulsado con un pulso de presión de 4,48 bar durante 0,8 min. El flujo de purga fue de 40 mL/min y la válvula estuvo cerrada durante 0,6 min. El volumen de inyección fue de 2 L y la temperatura del inyector fue de 256 ºC. La temperatura del detector μ-ECD fue de 310 °C con flujo de gas auxiliar (nitrógeno) de 10 mL/min. El detector NPD se trabajó a 330 °C con flujos constantes de gas auxiliar (nitrógeno), hidrógeno y aire de 10, 3 y 60 mL/min, respectivamente. El programa de temperatura del horno inició en 52 ºC (0 min), incrementándose la temperatura a una velocidad de 4 ºC/min hasta alcanzar 100 ºC, consecutivamente se aumentó la temperatura hasta 110 ºC a 2 ºC/min a continuación se incrementó hasta 130 ºC a una velocidad de 20 ºC/min, luego se llevó hasta 195 ºC a 4 ºC/min y finalmente se llegó a una temperatura de 280 ºC a una velocidad de 5 ºC/min. Se empeló un equipo para extracción en fase sólida marca Tecknokroma de 12 posiciones conectado a una bomba de vacío.
Metodologías de extracción de plaguicidas en aguas
Extracción líquido-líquido
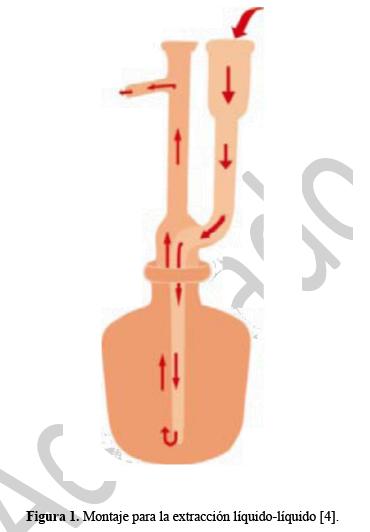
La extracción líquido – líquido se llevó a cabo usando n-hexano como solvente de extracción. 1L de agua se filtró a través de una membrana de 0,45 µm y se depositó en una botella ámbar, se adicionaron 10 g de cloruro de sodio y 60 mL de n- hexano. La botella tapada se colocó en agitación magnética durante 30 minutos, luego se realizó la recolección de la fase orgánica por medio del dispositivo que se muestra en la figura 1. La fase orgánica se filtró a través de 15 g de sulfato de sodio anhidro y se concentró a una temperatura de 35 ºC hasta aproximadamente 0,5 mL. El solvente se eliminó en su totalidad bajo una corriente de nitrógeno y el extracto se transfirió cuantitativamente con acetato de etilo a un balón aforado de 1 mL(7).
Extracción en fase sólida
La extracción en fase sólida se llevó a cabo empleando cartuchos LiChrolut® EN, previamente acondicionados con 3 mL de metanol y 3 mL de agua grado HPLC. 500 mL de agua previamente filtrada por una membrana de 0,45 μm se pasaron a través del cartucho a una velocidad de 15 mL/min aplicando vacío. Una vez se pasó la totalidad de la muestra, se
secó el cartucho durante 10 minutos en una corriente de nitrógeno. La elución de los analitos de llevó a cabo utilizando 6 mL de una mezcla acetato de etilo:metanol 1:1, la cual posteriormente se evaporó en su totalidad. El extracto se transfirió cuantitativamente a un balón aforado de 1 mL con acetato de etilo (20).
Aptitud y desempeño de las metodologías
Se llevaron a cabo ensayos de recuperación por triplicado con las mezclas de analitos, utilizando agua potable del grifo como matriz blanco, adicionando dicha mezcla a 1L o 500 mL de muestra, según la metodología. Como control de calidad, previo al procedimiento de extracción y análisis se adicionaron 20 μL de una mezcla de PCB 103 (2,2',4,5',6-pentaclorobifenil) y TPP (trifenil fosfato) denominados estándares subrogados. Además, al extracto final se le adicionaron 20 μL de una mezcla de PCB 52 (2,2',5,5'-tetraclorobifenil) y Sulfotep (ditiopirofosfato de O,O,O,O-tetraetilo), utilizados como estándares internos.
La aptitud de las metodologías se evaluó teniendo en cuenta los límites de detección y cuantificación, el porcentaje de recuperación y la precisión en condiciones de repetibilidad.
Límites de detección y cuantificación y selectividad
Se realizó la inyección de tres blancos de matriz, extraídos según las metodologías a evaluar en la misma secuencia con la mezcla de estándares preparada en extracto de matriz blanco. Se estimaron los límites de detección (LD) y cuantificación (LC) con base en el criterio de la EPA (21) Se realizó una inyección de blanco fortificado al valor del límite de detección y al límite de cuantificación estimados para verificar la capacidad instrumental de detectar el analito a estas concentraciones y garantizar la ausencia de interferencias en los tiempos de retención de los analitos de interés. Con base en éstos resultados se calcularon los límites de detección y cuantificación en matriz.
Porcentaje de recuperación y precisión
Se fortificaron por triplicado muestras blanco de matriz a las concentraciones establecidas como límite de cuantificación y se analizaron con cada una de las dos metodologías. Se determinó el porcentaje de recuperación y la desviación estándar relativa para cada compuesto.
Resultados y discusiòn
Con el fin de determinar los compuestos de interés a las concentraciones exigidas por la legislación colombiana (18), se calcularon los límites de detección y cuantificación de cada metodología. En la tabla 2 se observa la concentración máxima permitida por la normativa, así como los valores de límite de detección calculados.
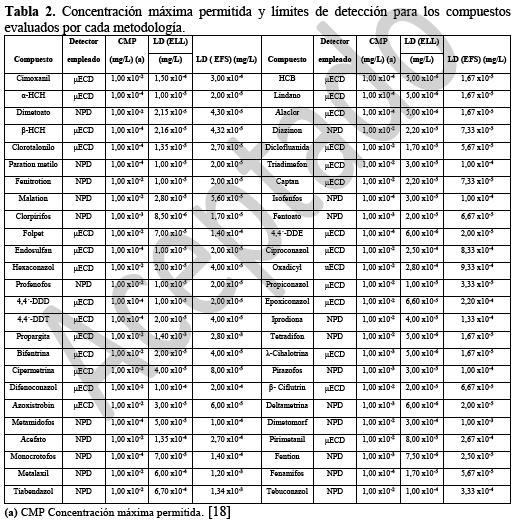
En esta tabla se muestra que para todos los compuestos se obtuvieron límites de detección por debajo de la concentración máxima permitida para las dos metodologías, a excepción del compuesto monocrotofos para el cual se obtuvo un límite superior a este valor en la metodología de extracción en fase sólida. En general, se observa que la mayoría de los compuestos tienen un límite de detección entre 1,00x10-05 mg/L y 1,00x10-04 mg/L, valor que se encuentra por debajo del límite más exigente de la normativa, el cual es 1,00x10-04 mg/L. Se determinó que utilizando la metodología por ELL se obtienen extractos con menos interferencias, lo cual incrementa la relación señal/ruido y permite obtener límites de detección menores a 1,00x10-05 mg/L para varios de los compuestos evaluados. Estos valores son comparables con los encontrados en el estudio de Majzik-Solymos et al. (5), en el que se empleó extracción líquido – líquido con un volumen de muestra superior (1L) y utilizando 200 mL diclorometano para la extracción de plaguicidas neutros y 300 mL de éter de petróleo para extraer compuestos ácidos. La metodología con extracción en fase sólida presentó extractos con mayores interferencias, por lo cual, los valores de límites de detección se ven incrementados para ciertos compuestos, como es el caso de monocrotofos, que aunque se observa por el detector NPD el cual presenta mayor selectividad, muestra interferencias que aumentan el ruido y por tanto el LD como se observa en la tabla 2. Los límites de detección obtenidos para este compuesto por las dos metodologías son en realidad bastante bajos, sin embargo empleando la metodología por EFS no se podría determinar la concentración exigida por la normativa colombiana.
Una vez establecido que los compuestos pueden ser determinados por las dos metodologías se evaluó la capacidad de estas para recuperar los analitos de estudio de la matriz de interés. Se realizaron ensayos de recuperación a una concentración equivalente al límite de cuantificación.
La extracción líquido – líquido es un método experimentalmente sencillo, económico y rápido de ejecutar, pues hace uso de las propiedades fisicoquímicas de los plaguicidas con baja solubilidad en agua y baja polaridad, para, por medio de una agitación magnética fuerte y constate durante 30 minutos, hacerlos pasar de la matriz acuosa al solvente de extracción, en este caso, n-hexano, aumentando la fuerza iónica con la adición de cloruro de sodio y facilitando así la partición.
Los criterios seleccionados para evaluar la recuperación de los compuestos fueron los establecidos en el documento SANCO (22) que, aunque establece los lineamientos para la determinación de residuos de plaguicidas en alimentos y piensos, pueden ser extrapolados para la determinación en otras matrices de interés.
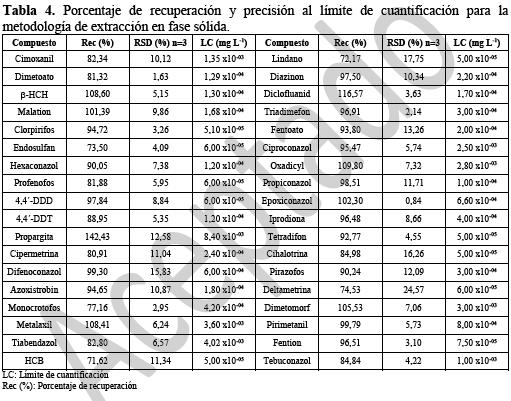
En la tabla 3 se muestran los porcentajes de recuperación, límites de cuantificación y desviación estándar relativa empleando la metodología ELL para los compuestos que cumplieron los criterios establecidos en SANCO.
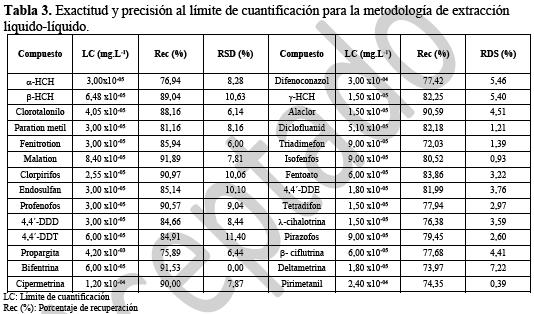
Se observa que los compuestos recuperados corresponden a los plaguicidas organoclorados y piretroides incluidos en el ensayo, así como algunos organofosforados tales como el malation, el clorpirifos y benceno sustituidos como el clorotalonilo entre otros, que por su elevado coeficiente de partición octanol/agua (23) pasan fácilmente a la fase orgánica, obteniendo así altos porcentajes de recuperación. Compuestos de la familia de los azoles, como el difenoconazol o epoxiconazol, y organofosforados de mayor polaridad como el dimetoato y el monocrotofos tienen porcentajes de recuperación muy bajos o nulos. Esto se debe a su elevada solubilidad en agua y bajo coeficiente de partición octanol/agua, que en algunos casos es menor a la unidad (Monocrotofos=0,6) (23).Se observa también que para todos los plaguicidas recuperados se tienen desviaciones estándar relativas dentro del rango esperado (<20% (22)) haciendo adecuada la metodología para su análisis.
En la Figura 2a se puede ver que empleando ELL, el 56% de los compuestos evaluados presentan un porcentaje de recuperación entre 70% y 120% mientras que el 26% tienen una recuperación nula; el porcentaje restante presenta recuperación entre el 10% y el 70%.
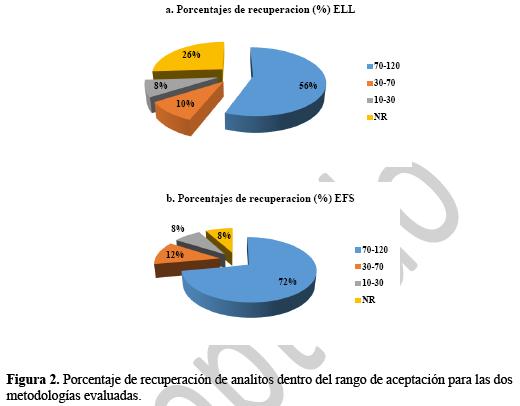
En cuanto a la metodología de extracción en fase sólida, en la figura 2b se observa que un 72% de los compuestos evaluados presentaron porcentajes de recuperación dentro del rango de aceptación de la metodología (70-120%) (22) mientras que un 8% presentó recuperación nula. Estos resultados son comparables con los obtenidos por Robles-Molina et al. (5), quien en su estudio comparativo determinó que, a una concentración de fortificación de 1,5x10-4 mg/L, de un total de 57 compuestos de diferentes familias químicas, un 53% presentaban porcentajes de recuperación superiores al 70% empleando ELL y un 63% empleando EFS.
El proceso de extracción en fase solida tiene como objetivo realizar el aislamiento selectivo de los analitos objetivo en una muestra compleja y al mismo tiempo realizar un proceso de concentración, como es el caso del presente estudio. En el proceso se emplean interacciones reversibles, entre el analito y la superficie de la fase estacionaria. Comercialmente se encuentran diversas modificaciones en materia de adsorbentes; en este caso se utilizó una fase polimérica compuesta por un polímero de etilvinilbenceno y divinilbenceno entrecruzado (20), el cual provee una gran área superficial y permite retener sustancias de mayor polaridad que pueden ser eluidas posteriormente utilizando la mezcla metanol:acetato de etilo. Debido a la polaridad intermedia de la fase estacionaria, compuestos como el acefato (Log Kow=-0,89) y su metabolito, el metamidofos (Log Kow=-0,89) que poseen muy elevada solubilidad en agua (650 g/L a 25°C y >2000 g/L a 25°C respectivamente) (24) no se quedan retenidos en la fase estacionaria, sino que tienden a quedarse disueltos en la matriz acuosa, por lo tanto su recuperación es baja o nula. Este tipo de compuestos requiere el uso de fases estacionarias con sustituyentes polares que permitan su retención y elución selectiva con disolventes de elevada polaridad. Así mismo, compuestos como algunos organoclorados o como el clorotalonilo, que es un benceno sustituido no fueron eluidos con la mezcla de solventes empleada, quedándose retenidos en la superficie del adsorbente, por lo que su recuperación en EFS es muy baja o nula. En la tabla 4 se muestran los porcentajes de recuperación, los límites de cuantificación y la desviación estándar relativa para los compuestos extraídos por esta metodología. En su estudio Robles-Molina et al. (5) muestran que pueden encontrarse marcadas diferencias en la recuperación de analitos de diferentes familias químicas empleando una u otra técnica, lo cual se evidenció con las triazinas, que presentaron porcentajes de recuperación superiores al 90% con EFS y menores a 60% con ELL. Robles-Molina et al. (5), también determinaron que los compuestos organoclorados presentaron mejores recuperaciones con ELL y que para los organofosforados se obtienen, en general, buenos porcentajes de recuperación con ambas metodologías.
Se evidenció que 22 de los 50 plaguicidas presentaron recuperación entre el 70% y el 120% tanto por EFS como por ELL. Compuestos organoclorados como el 4,4´-DDD, el endosulfan y el alaclor tienen porcentajes de recuperación estadísticamente superiores empleando EFS (t student p<0,05), sin embargo la metodología de ELL es más adecuada para su análisis puesto que el valor de RSD es menor empleando ésta metodología. De los 22 compuestos, 13 no presentaron diferencias significativas (p>0,05) en la recuperación por las dos metodologías.
Conclusiones
En el presente estudio se realizó la comparación del desempeño en cuanto a detección, porcentaje de recuperación y precisión de dos metodologías para la extracción de 50 plaguicidas en muestras de agua potable empleando cromatografía de gases acoplada a detector de micro captura electrónica y de nitrógeno fosforo. Las dos metodologías permiten determinar los analitos evaluados de acuerdo a los valores establecidos por la normativa en Colombia, con excepción del insecticida monocrotofos, cuyo límite de detección es superior al exigido.
Se determinó que 36 de los 50 compuestos evaluados pueden ser determinados empleando la metodología de extracción en fase sólida mientras que empleando extracción líquido-líquido pueden analizarse 28 de ellos. Así mismo, se determinó que 22 de los compuestos evaluados pueden ser determinados con cualquiera de las dos metodologías, teniendo porcentajes de recuperación mayores con la metodología de extracción en fase sólida y siendo más precisa la extracción líquido -líquido. Se determinó que el insecticida acefato y su metabolito metamidofos no pueden ser analizados por ninguna de las dos metodologías propuestas debido a su elevada polaridad y solubilidad en agua.
Agradecimientos
Expresamos nuestros agradecimientos al Departamento Administrativo de Ciencia, Tecnología e Innovación, COLCIENCIAS y al Laboratorio de Análisis de Residuos de Plaguicidas LARP del Departamento de Química de la Universidad Nacional de Colombia, sede Bogotá por la financiación de este proyecto.
Referencias
1. Estévez-Arias, M.; López-Periago E.; Martínez-Carballo, E.; Simal-Gándara, J.; Mejuto, J-C.; García Río, J. The mobility and degradation of pesticides in soils and the pollution of groundwater resources. Agriculture, Ecosystems & Environment. 2008. 123(4): 247-260. [ Links ]
2. Chowdhary, S., R. Bhattacharyya, and D. Banerjee, Acute organophosphorus poisoning.Clinica Chimica Acta, 2014. 431: 66-76. [ Links ]
3. Environmental Protection Agency. Methods for organic chemical analysis of municipal and industrial wastewater. Method 608-organochlorine pesticides and PCBs. 2007. [ Links ]
4. Wu, J.; Lu, J.; Wilson, C. Effective liquid-liquid extraction method for analysis of pyrethroid and phenylpyrazole pesticides in emulsion-prone surface water samples. Journal of Chromatography A, 2010. 1217. 41: 6327-6333. [ Links ]
5. Robles-Molina, J.; López, G.; García-Reyes, JF., Molina-Díaz, A. Comparative evaluation of liquid-liquid extraction, solid-phase extraction and solid-phase microextraction for the gas chromatography-mass spectrometry determination of multiclass priority organic contaminants in wastewater. Talanta. 2013. 117: 382-391. [ Links ]
6. Majzik-Solymos, E.; Visi, E.; Károly G.; Beke-Berczi, B.; Györfi, L. Comparison of extraction methods to monitor pesticide residues in surface water. J Chromatogr Sci. 2001. 39: 325-31. [ Links ]
7. Mojica, A. Determinación de residuos de plaguicidas en aguas y suelos de la región del lago de Tota. Tesis de Maestría. Departamento de Química. 2010, Universidad Nacional de Colombia. [ Links ]
8. Tankiewicz, M.; Fenik, J.; Biziuk, M.; Solventless and solvent-minimized sample preparation techniques for determining currently used pesticides in water samples: A review. Talanta. 2011. 86: 8-22. [ Links ]
9. Seebunrueng, K.; Santaladchaiyakit, Y.; Srijaranai, S. Vortex-assisted low density solvent based demulsified dispersive liquid-liquid microextraction and high-performance liquid chromatography for the determination of organophosphorus pesticides in water samples. Chemosphere. 2014. 103: 51-58. [ Links ]
10. Farajzadeh, M.A.; Khoshmaram, L.; Air-assisted liquid-liquid microextraction-gas chromatography-flame ionisation detection: A fast and simple method for the assessment of triazole pesticides residues in surface water, cucumber, tomato and grape juices samples. Food Chemistry. 2013. 141(3): 1881-1887. [ Links ]
11. Jiménez, J.J. Simultaneous liquid-liquid extraction and dispersive solid-phase extraction as a sample preparation method to determine acidic contaminants in river water by gas chromatography/mass spectrometry. Talanta. 2013. 116: 678-687. [ Links ]
12. Żwir-Ferenc, A.; Biziuk, M. Solid Phase Extraction Technique - Trends, Opportunities and Applications. Polish J. of Environ. Stud. 2006. 15(5): 13-17. [ Links ]
13. Latrous EI Atrache, L.; Ben Sqhaier, R.; Bejaoui Kefi, B.; Haldys, V.; Tortajada, J. Factorial design optimization of experimental variables in preconcentration of carbamates pesticides in water samples using solid phase extraction and liquid chromatography-electrospray-mass spectrometry determination. Talanta, 2013. 117: 392-398. [ Links ]
14. Hurtado-Sánchez, M.C.; Romero-Gónzalez, R.; Rodríguez Cáceres, MI.; Durán-Merás, I.; Frenich, AG. Rapid and sensitive on-line solid phase extraction-ultra high performance liquid chromatography-electrospray-tandem mass spectrometry analysis of pesticides in surface waters. Journal of Chromatography A. 2013. 130: 193-202. [ Links ]
15. Patsias, J.; Papadopoulou-Mourkidou, E. Rapid method for the analysis of a variety of chemical classes of pesticides in surface and ground waters by off-line solid-phase extraction and gas chromatography-ion trap mass spectrometry. Journal of Chromatography A. 1996. 740(1): 83-98. [ Links ]
16. Masiá, A.; Ibáñez, M.; Sancho, JV.; Picó Y.; Hernández, F. Combined use of liquid chromatography triple quadrupole mass spectrometry and liquid chromatography quadrupole time-of-flight mass spectrometry in systematic screening of pesticides and other contaminants in water samples. Analytica Chimica Acta. 2013. 761: 117-127. [ Links ]
17. Rodríguez, H.A.; Guerrero, J.; Castro, R. Determinación de residuos de glifosato y de su metabolito ácido aminometilfosfonico en aguas mediante cromatografia liquida de alta eficiencia con derivacion poscolumna y deteccion por fluorescencia. Revista Colombiana de Química. 2002. 31(1): 7 - 18. [ Links ]
18. Ministerio de la protección social, Ministerio de ambiente, vivienda y desarrollo territorial. Resolución 2115 del 22 de Junio de 2007. Capítulo 8. [ Links ]
19. World Healt Organization - International agency for cancer research. Agents classified by the IARC monographs, volumes 1-109. 345 Disponible en http://monographs.iarc.fr/ENG/Classification/. [consultado el 10 de Julio de 2014] [ Links ].
20. Junker-Buchheit, A.; Witzenbacher, M. Pesticide monitoring of drinking water with the help of solid-phase extraction and high-performance liquid chromatography. Journal of Chromatography A. 1996. 737(1): 67-74. [ Links ]
21. Corley, J. Best practices in establishing detection and quantification limits for pesticide residues in foods. En: Handbook of Residue Analytical Methods for Agrochemicals. J.W.S. Ltd. Editor. 2003. [ Links ]
22. SANCO. Method Validation and quality Control Procedures for pesticide residues analysis in food and feed. 2011. Disponible en http://ec.europa.eu/food/plant/protection/resources/qualcontrol_en.pdf. [Consultado el 10 de Julio de 2014] [ Links ].
23. Kegley, S.E.; Hill, B.R.; Orme S.; Choi A.H. PAN Pesticide Database, Pesticide Action Network. Estados Unidos. 2014. Disponible en: http://www.pesticideinfo.org. [Consultado el 25 de junio de 2014] [ Links ].
24. Yen, J.H.; Lin, KH.; Wang, Y.S. Potential of the insecticides acephate and methamidophos to contaminate groundwater.Ecotoxicol Environ Saf. 2000. 45(1): 79-86. [ Links ]
