











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
En las últimas décadas, los polímeros conductores han tenido gran relevancia debido a sus múltiples aplicaciones gracias a que presentan propiedades como una gran estabilidad térmica y la capacidad de formar películas [1]. Estas ventajas facilitan el procesamiento y la fabricación de los polímeros a costos bastante razonables [2]. El uso de heterociclos alternados de cinco miembros (furano, tiofeno, pirrol, borol) mejora de forma significativa las características conductoras de los polímeros, basándose, principalmente, en una disminución de la energía entre el orbital molecular ocupado de más alta energía (HOMO) y el orbital molecular desocupado de más baja energía (LUMO) [3]. El diseño de mejores materiales electrónicos orgánicos requiere una comprensión profunda de la estructura electrónica de los polímeros conjugados y de los factores que afectan su estabilidad y reactividad.
Ha habido un gran interés en estudiar, teóricamente, los aspectos estructurales y electrónicos que influyen en la conductividad de una serie de copolímeros de furano-tiofeno monosustituidos, pues es posible observar y cuantificar el efecto en el comportamiento de las propiedades electrónicas de la conductividad, cuando se utilizan grupos sustituyentes, tales como: carboxilo (COOH), metilo (Me), hidroxilo (OH), ciano (CN) y fluoruro (F), como sustituyentes en el carbono C3 y C10 de cada anillo. Cada heterociclo sustituido muestra un comportamiento químico diferente al de sus respectivos polímeros, lo cual lleva a preguntarse acerca de qué tan buen electroconductor puede ser un copolímero formado por diferentes unidades estructurales de materiales poliméricos [4]. La exploración del efecto de los diferentes tipos de grupos sustituyentes sobre la conductividad en copolímeros de furano-tiofeno puede ser analizada, aprovechando la diferencia energética entre las bandas de valencia y las bandas de conducción de un polímero (en inglés band gap) [5]. La energía del band gap (Eg) es calculada de forma análoga a los cálculos en estado sólido de conductividad, es decir, hallando la diferencia entre la energía del HOMO y el LUMO [6].
En química computacional, el uso del DFT ha aumentado progresivamente, pues este resulta de gran importancia para los cálculos que implican sistemas finitos, en la medida en que incluye una correlación electrónica, ya que asume que la energía del estado fundamental de un sistema electrónico está únicamente definida por su densidad electrónica p0(x, y, z) [7]. Los métodos DFT han sido utilizados para hacer buenas predicciones de la diferencia energética entre los orbitales HOMO y LUMO en polímeros orgánicos conjugados. Si bien aún existe una controversia en torno a la interpretación de la energía de los orbitales, se ha encontrado que la teoría DFT ofrece una buena estimación del BG [8]. El criterio para escoger los sustituyentes mencionados se basa en la naturaleza electrodonora de los sustituyentes, lo cual posiblemente facilita la reactividad de los anillos aromáticos. En este estudio se emplea la extrapolación a cadena de longitud infinita con el fin de conocer algunas propiedades del polímero. Esta metodología es fiable, ya que, para la mayoría de los polímeros, las propiedades de sus oligómeros se encuentran bien determinadas [9]. Así pues, el objetivo de este trabajo es comparar los efectos que tienen los diferentes sustituyentes en la cadena oligomérica sobre las propiedades de conducción eléctrica del material de copolímero de furano-tiofeno.
Materiales y métodos
Detalles computacionales
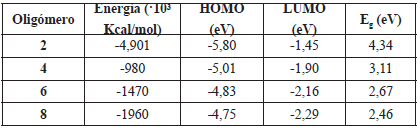
Todos los cálculos de los oligómeros (N = 2→8) realizados en este trabajo se hicieron con la ayuda del paquete de programas Gaussian 09 [10].
Las geometrías de los oligómeros neutros y cargados que contienen 2, 4, 6 y 8 anillos fueron optimizadas a nivel DFT, utilizando el funcional híbrido B3LYP [10], junto con el conjunto de base 6-311G* [11].
La geometría de los monómeros de furano y tiofeno y los monómeros de furano y tiofeno sustituidos con grupos metilo, hidroxilo, ciano, carboxilo y átomos de flúor (Figura 1) se optimizó en estados neutros y cargados al mismo nivel de teoría descrito anteriormente.
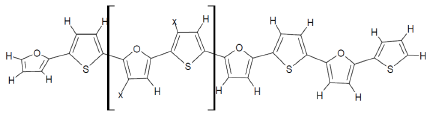
Figura 1 Estructura molecular de los oligómeros de furano-tiofeno estudiados (donde X=Me, COOH, OH, CN y F). Me=metilo, COOH=carboxilo, OH=hidroxilo, CN=ciano y F=átomos de flúor.
Los potenciales de ionización (PI) se calcularon como diferencias de energías entre el catión y la molécula neutra. Estos potenciales de ionización fueron calculados a partir de las geometrías en estado fundamental optimizada [12]. Los cálculos fueron realizados sobre los oligómeros que contienen 2, 4, 6 y 8 anillos sustituidos con los grupos Me, CN, COOH, OH y átomos de F. Los parámetros de polímero (PI y Eg) fueron obtenidos por la extrapolación lineal de la propiedad de interés versus el inverso del número de monómeros (1/N) y por la extrapolación a cadena de longitud infinita (1/N=0) [13].
También, se realizaron extrapolaciones a la longitud de la cadena infinita para los polímeros con estados cargados con el fin de evaluar el valor de Eg. Además de estos cálculos, se obtuvo una visualización del orbital molecular desocupado de más baja energía.
Resultados y discusión
En la Tabla 1 se muestran los valores de los potenciales de ionización, todos los cálculos se realizaron utilizando la diferencia entre la energía de la molécula cargada (catión) y la molécula en el estado neutro. Los polímeros están abreviados como sigue: copolímero furano-tiofeno (COFT), copolímero furano-tiofeno sustituido con flúor (COFT-F), copolímero furano-tiofeno sustituido con metilo (COFT-Me), copolímero de furano-tiofeno sustituido con carboxilo (COFT-COOH), copolímero furano-tiofeno sustituido con ciano (COFT-CN) y copolímero furano-tiofeno sustituido con hidroxilo (COFT-OH).
Tabla 1 Potencial de ionización (PI) de copolímeros de furano-tiofeno y copolímeros de furano-tiofeno sustituidos, calculados a nivel B3LYP /6-311G* en electrón voltios (eV).
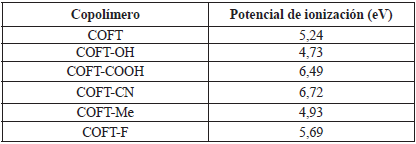
Los resultados mostrados en la Tabla 1 muestran la siguiente tendencia:
COFT-CN ˃ COFT-COOH ˃ COFT-F ˃ COFT ˃ COFT-Me ˃ COFT-OH
Lo anterior está en consonancia con la naturaleza electrodonora de los diferentes sustituyentes que alteran el potencial de ionización del copolímero. Dentro de este grupo de sustituyentes y debido a la naturaleza química de estos, se destaca el grupo CN, el cual es un fuerte desactivador del anillo aromático. En general, el COFT-CN presenta el mayor potencial de ionización, lo cual indica que es más difícil extraer un electrón de esta estructura molecular.
En el otro extremo de los copolímeros estudiados, el copolímero que presenta menor potencial de ionización es aquel que fue sustituido con el grupo OH, el cual es un activante muy fuerte de los anillos aromáticos, debido a la inducción y a la electronegatividad del oxígeno. Sin embargo, el efecto inductivo electroatractor es superado por un efecto donor mucho más importante que involucra los electrones no apareados del oxígeno [14], y tal relación hace que esta molécula muestre un menor valor de potencial de ionización.
De acuerdo con la tendencia anterior, se puede observar que el COFT-Me tiene un valor de potencial de ionización menor que el copolímero sin sustituyentes (COFT). Lo anterior se debe a que el grupo Me es un grupo donor de electrones [15], mientras que el grupo COOH y el átomo de F son atractores de electrones [15], los cuales son capaces de retenerlos en su estructura, dificultando la formación de cationes en este tipo de polímeros. En la Tabla 2 se muestran los valores de Eg para los oligómeros de furano-tiofeno y los oligómeros de furano-tiofeno sustituidos.
Tabla 2 Valores de Eg en electrón voltios (eV) extrapolado a cadena de longitud infinita, usando el nivel de cálculo DFT-B3LYP-6311G*.
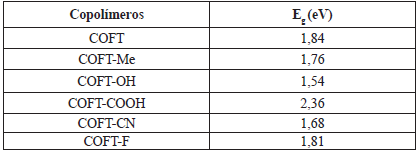
En términos generales los copolímeros sustituidos presentan un menor valor de Eg respecto al copolímero no sustituido. Este comportamiento, también, es mostrado por el polifurano sustituido (ver Tabla 2) [16]. Cabe indicar que el COFT-COOH no sigue esta tendencia de comportamiento, lo cual puede ser explicado por problemas de efectos estéricos que hacen que la molécula pierda planaridad y, por ende, que el traslape de los orbitales no sea el más óptimo, provocando así que la conductividad disminuya. El estudio muestra, además, que el COFT-OH presenta las mejores características electroconductoras. La tendencia en orden decreciente de band gap es la siguiente:
COFT-COOH > COFT > COFT-F > COFT-CN > COFT-Me > COFT-OH
Esta tendencia indica que el grupo OH, al ser un fuerte activador de los anillos aromáticos [17], permite la modificación de la diferencia energética entre los orbitales moleculares HOMO y LUMO al alterar la densidad electrónica de este último [18]. Este cambio facilita la probabilidad de que un electrón sufra un decaimiento de energía al orbital LUMO, estrechando así la diferencia de energía entre estos orbitales, lo cual da como resultado un valor menor de Eg, esperando así que el copolímero presente mejores características electroconductoras de acuerdo a este criterio.
Por otra parte, el COFT-COOH, los cuales son fuertes desactivantes de los anillos aromáticos [18], muestra un incremento en el valor de Eg. Este análisis se interpreta de manera más sencilla al graficar como ejemplo la correlacion lineal de Eg versus 1/N del oligómero de COPFT-H (Figura 2). En los cálculos realizados se encontró que no había diferencia significativa entre los valores de Eg de los copolímeros en estado neutro y los cationes respectivos; mientras que los copolímeros cargados negativamente presentaron menores valores de Eg respecto a sus estados neutros.
Se realizó un análisis donde se escogió un segmento de la molécula que reprodujo el comportamiento de esta, y se examinó la contribución de cada átomo al LUMO, para corroborar así las observaciones anteriormente mencionadas.
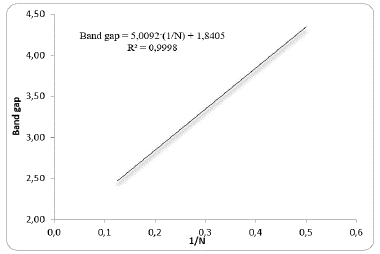
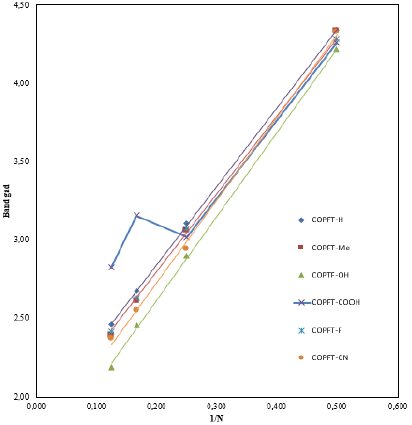
La correlacion lineal de COPFT-H es casi del 100%, obteniendo un acoplamiento lineal de 99,9% de correlacion, lo cual demuestra que el modelo de cálculo por cada copolímero se ajusta casi de forma ideal. Tambien, se hizo de manera identica para cada sustituyente, obteniendo así la siguiente Figura 3.
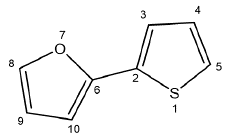
Para el análisis de las contribuciones del orbital molecular LUMO se tomó un segmento de los diferentes oligómeros como se muestra en la Figura 4. Es decir, se tomó aquella parte de la molécula que conserva cierta simetría en cuanto a propiedades electrónicas se refiere, pues si la cadena oligomérica crece a ambos extremos habrá una región molecular que conserve ciertas propiedades. Lo anterior es posible en la medida en que los sustituyentes se encuentran en posiciones opuestas y no afectarán la regioisomería del oligómero, ya que no son sustituyentes vecinos y no generarán impedimentos estéricos, manteniendo así las propiedades optoelectrónicas [19]. Por esta razón, se tomó la región central de los copolímeros de furano-tiofeno.
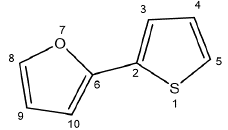
Figura 4. Segmento tomado para el análisis de la contribución de cada átomo al LUMO.
Las sustituciones a la cadena polimérica se realizaron sobre las posiciones 3 y 10 del segmento del copolímero como se observa en las Figuras 3 y 4. La elección de estas posiciones para ser sustituciones se realizó con el fin de que los polímeros fueran simétricos [19].
Otro aspecto a tener en cuenta para el análisis de la electroconductividad fue la contribución de cada átomo al LUMO, este orbital es el primero que se encuentra disponible para recibir electrones, y, de acuerdo a la teoría de bandas, esta sería la banda de conducción. Este aspecto es muy importante, ya que una buena contribución de cada átomo a este orbital permitirá a los electrones fluir de una manera más óptima por la estructura del polímero [20].
La Figura 5 muestra la contribución de cada átomo del segmento al LUMO. En esta gráfica se observan las variaciones más relevantes sobre los carbonos por donde crece la cadena (carbonos 5 y 8) y los que se encuentran sustituidos. Tal comportamiento era esperado, pues en esta contribución existe la modificación de las probabilidades de encontrar un electrón al estar presente otro tipo de nube electrónica por efecto de los sustituyentes.
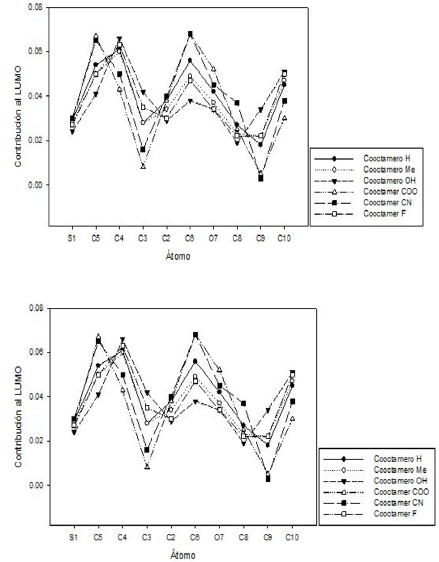
Figura 5 Contribución de los átomos al LUMO de las moléculas optimizadas al nivel DFT B3LYP 6-311G*.
Aquí, se compara el comportamiento del copolímero sustituido con hidroxilo (mejor electroconductor) y el sustituido con grupos carboxilo (peor electroconductor), lo cual permite observar que el oligómero sustituido con grupos hidroxilo presenta un comportamiento más armónico, en cuanto a la contribución de los átomos al LUMO se refiere.
A su vez, dicha contribución aumenta y disminuye de forma lenta, lo cual evidencia que existe una mejor distribución de la densidad electrónica para ubicar un electrón, en el caso que este pase a la banda de conducción. Por el contrario, el oligómero sustituido con grupos carboxilos presenta altibajos muy pronunciados que posiblemente no favorecerán la resonancia ni el posible transito simple de los electrones.
Para el caso del copolímero sin sustituir no se muestra una distribución tan armónica como en el caso del sustituido con hidroxilo, pero tampoco presenta cambios tan marcados como el presentado en el caso del oligómero sustituido con carboxilo, por tanto, se aprecia que las contribuciones al orbital LUMO dan una idea del camino que puede seguir el flujo electrónico y sobre qué átomos se ven favorecidas y desfavorecidas tales contribuciones, las cuales son de gran utilidad para inferir el comportamiento electroconductor de este tipo de moléculas [21].
Conclusiones
En la presente investigación se estudiaron las propiedades electrónicas de transición e ionización de una serie de copolímeros de furano-tiofeno y copolímeros de furano-tiofeno sustituidos, empleando desde dímeros a octámeros y extrapolando las propiedades a cadena de longitud infinita.
El análisis de las propiedades electrónicas muestra que los copolímeros sustituidos con grupos hidroxilos presentan el mejor Eg de todas las especies estudiadas, pues al realizar la extrapolación a cadena de longitud infinita dicho valor se ubica en un rango menor a los presentados por los otros copolímeros estudiados. Este hecho, también, es corroborado, porque se presenta una mejor contribución de los átomos del segmento al LUMO, lo cual indica que el traslape de estos orbitales es más óptimo.
Los copolímeros sustituidos con grupos carboxilos presentaron las propiedades electroconductoras más bajas entre las especies estudiadas, este hecho puede estar relacionado con problemas de efectos estéricos, provocando una desfavorabilidad en el traslape adecuado de los orbitales moleculares, lo cual implica que haya un alto valor de Eg y que la contribución de los átomos del segmento del copolímero al LUMO no sea la más óptima para favorecer la conductividad eléctrica.

