











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroducción
Existen reportes históricos desde tiempos inmemorables de muchas clases de pandemias que han azotado a la humanidad y que han dejado como consecuencia una serie de disrupciones en la cotidianidad de las personas, además de muerte y desolación. Particularmente, en los últimos años y en especial desde casi del principio del siglo XXI ha habido una serie de pandemias relacionadas con virus [1,2]. A la fecha, vivimos una pandemia ocasionada por el virus del SARS-CoV-2, un virus perteneciente a la familia de los coronavirus y causante de la enfermedad COVID-19, situación que ha conllevado a un despliegue interdisciplinar para la búsqueda de vacunas y tratamiento de la enfermedad [3,4].
Una infección muy parecida a la que vivimos actualmente, pero con menores consecuencias es la gripa aviar, que tuvo unos orígenes muy parecidos a la COVID-19 [5]. La gripe aviar es una infección causada por el virus de la influenza aviar o virus de la influenza A (H5N1); este microorganismo se presenta de forma natural en las aves y es capaz de infectar una variedad de animales incluyendo humanos, cerdos, caballos, mamíferos marinos y por supuesto aves [6]. Los virus de la influenza pertenecen a la familia Orthomyxoviridae y se distribuyen en tres géneros: influenza virus A, influenza virus B e influenza virus C, que corresponden a los virus de influenza tipo A, B y C, respectivamente. La diferencia principal entre los géneros radica en las variaciones antigénicas en la proteína de la matriz y de la nucleoproteína que se utilizan para la caracterización del virus y que son específicas para cada género [6]. El genoma de los virus A está formado por 8 piezas de RNA de cadena sencilla y polaridad negativa, es decir, no pueden ser traducidos directamente a la proteína. Las secuencias de los extremos de cada RNA están conservadas entre sí entre todos los virus tipo A, y son parcialmente complementarias. Cada segmento de RNA está asociado a la polimerasa viral, que es un complejo de tres subunidades distintas, PB1, PB2 y PA, mediante interacción con los extremos del RNA y está protegido por asociación a monómeros de la nucleoproteína (NP), uno de los componentes mayoritarios en el virión [7].
En la superficie del virión, e insertadas en su membrana, se encuentran dos glicoproteínas, la hemaglutinina (HA) y la neuraminidasa (NA), cuya estructura determina los subtipos serológicos que podemos definir entre los virus gripales tipo A. Así, existen 16 subtipos diferentes de HA (H1-H16) y 9 de NA (N1-N9). Además, en el virión existen cantidades pequeñas de otra proteína denominada M2, que constituye un canal iónico dependiente del pH del medio y NEP, que está implicada en la exportación de vRNPs del núcleo [7]. Además de las proteínas citadas, los virus gripales tipo A expresan otras proteínas importantes para su replicación o su interacción con el huésped, que no están presentes en la partícula viral; la proteína NS1 une RNA de doble cadena, modula la replicación viral y es esencial para bloquear la respuesta celular a la infección, que está mediada por interferón [8]. La proteína PB1-F2 parece modular la apoptosis o muerte celular programada como consecuencia de la infección [9].
La HA es el mayor determinante antigénico de los virus de la influenza A y B, al cual están dirigidos los anticuerpos neutralizantes, por lo tanto, es el componente crucial de las vacunas actuales utilizadas para el control de la influenza. Esta proteína, una vez activada de manera enzimática por proteasas del tracto respiratorio, es la responsable de la unión del virus a sus receptores celulares de ácido siálico (Neu5Ac) y de la fusión de la envoltura viral con la membrana citoplasmática de la célula diana, procesos que determinarán la penetración del núcleo cápsula en el interior celular [10,11]. Generalmente, el virus de la influenza aviar (AI) no infecta a los humanos, debido a la respuesta primaria que presentan a la infección, como los receptores celulares específicos. No obstante, el virus puede cruzar ocasionalmente las barreras de la especie e infectar directamente a los humanos, incluyendo a las cepas altamente patogénicas que pueden causar una enfermedad fatal [12,13,14]. El subtipo H5N1 tiene la capacidad de cruzar la barrera entre las especies aviar y humana, por lo que este virus es considerado potencialmente pandémico; sus ancestros son virus aviares altamente patógenos encontrados previamente en pollos, gansos y patos de China [15]. De ahí, la necesidad de la búsqueda de moléculas inhibidoras para la neuraminidasa de este virus. Por otra parte, autores como Miki y Col (2007), han evaluado la actividad inhibitoria de flavonoides sobre la neuraminidasa de H1N1, debido a que estos son capaces de unirse a dicha proteína [16,17,18]. En este sentido, este estudio tiene por objetivo realizar el acoplamiento molecular y modelado tridimensional por homología de flavonoides derivados de amentoflavona con las neuraminidasas H1N1 y H5N1 del virus de gripe aviar. Además, cabe señalar que la metodología usada en el presente estudio responde a la serie de preguntas planteadas en esta investigación [19-22].
Materiales y métodos
Conjunto de estudio y diseño de estructuras
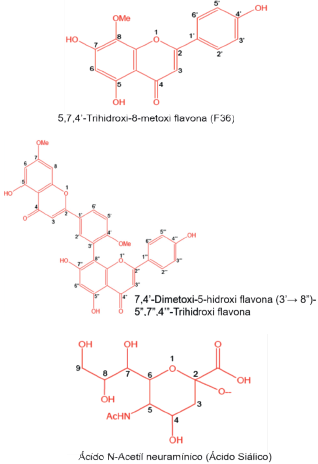
Para este trabajo se emplearon como moléculas base biflavonoides derivados de la amentoflavona, tomadas de los trabajos reportados por [23,24], quienes emplearon Ginkgo biloba L. y Cephalotaxus harringtonia K. para su extracción. Seguido, las estructuras fueron dibujadas y diferentes configuraciones relativas R y S fueron obtenidas para aquellas moléculas sustituidas con ácido siálico (Gauss View 3.09). Con el propósito de obtener las moléculas energéticamente favorables se realizó una optimización empleando el método semiempírico AMP, incorporado en el programa Gaussian 09. Las ocho moléculas seleccionadas se muestran en la Figura 1 y en la Tabla 1 se indican sus actividades inhibitorias expresadas como IC50 para la neuraminidasa de la cepa A/PR/8/34 (H1N1) [17].
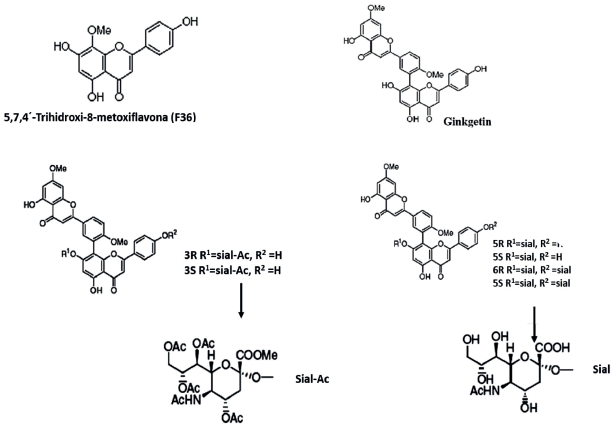
Obtención de estructuras tridimensionales de las proteínas H5N1 y H1N1
Inicialmente se realizó una búsqueda exhaustiva de las proteínas de interés en la Protein Data Bank (PDB), obteniendo así la estructura cristalográfica de la proteína A/Vietnam/1203/04 (H5N1) con código 2HTY y resolución de 2,5 A [25]. Sin embargo, para la cepa A/PR/8/34 (H1N1) no se encontró una estructura cristalográfica disponible, razón por la cual se hizo necesario predecir la estructura 3D mediante la realización de un modelo tridimensional teórico. Para esto se obtuvo la estructura primaria en formato FASTA de la neuraminidasa del virus de influenza A (H1N1) en la base de datos de la NCBI, código de acceso NP_040981 [26]. En seguida, se hizo la búsqueda de homólogos en la PDB usando el algoritmo de alineamiento BLAST [27] y posterior alineamiento secuencial con el servidor Clustal W [28]. Las estructuras secundarias fueron predichas con servidor Sympred [28] y la estructura 3D de H1N1 se obtuvo con los servidores de modelado por homología Swiss Model, Geno 3D y 3D-Jigsaw. También, se calculó el RMSD de los modelos obtenidos empleando el programa Swiss.pdb.Viewer (Deep View), opción Magic Fit, en la cual los carbonos alfa de los modelos son superpuestos con los de la estructura molde.
Para la optimización del modelo elegido se utilizó el campo de fuerza Tripos, con la asignación de las cargas de Gasteiger-Marsili en gradiente conjugado disponible en el módulo de composer del paquete de programa Sybyl 7.3. El solvente se adicionó de manera implícita (constante dieléctrica de 78) y los todos los hidrógenos fueron añadidos a la estructura. Por último, la evaluación de la estructura del modelo se realizó teniendo en cuenta tres componentes: 1) Potencial (servidores Eval23D, Verify3D, Prosa II y EvTree); 2) estereoquímica (servidor imoltalk) y 3) normalidad (servidor ProSA- Web).
Acoplamiento molecular de H1N1 y los ligandos biflavonoides El acoplamiento molecular entre la proteína H1N1 seleccionada y los ligandos bajo estudio se realizó en el programa FlexX incorporado en SYBYL 7.3. Para esto, se extrajo una porción de la proteína la cual incluyó el sitio activo y residuos de aminoácidos cercanos a este en un radio de 6,5Á. Los puentes de hidrogeno seleccionados estuvieron en el rango de 2,7 a 3,1Á y ángulos entre 120°-180°, debido a que son valores favorables [29].
El análisis de las interacciones se llevó a cabo con el servidor LPC/ SCU y para la visualización del complejo se empleó el programa Pymol. Para cada uno de los cálculos se realizó el análisis estadístico denominado Rank by Rank (Total store, D-Score, G-Score, PMF-Score y CHEM-Score), el cual permitió escoger el complejo energéticamente más favorable y, por tanto, la mejor pose para cada ligando.
Acoplamiento molecular entre H5N1 NA y los ligandos biflavonoides (diferente proteína)
A diferencia del acoplamiento molecular con H1N1, el estudio proteína-ligando con H5N1 NA se llevó a cabo con cuatro ligandos, debido a que solo tres de ellos mostraron alta potencia inhibidora (F36, 5R, 6R) y en el caso de Ginkgetin, a pesar de no ser un buen inhibidor, fue seleccionado como elemento de comparación.
Resultados y discusión
Obtención de la estructura tridimensional de la neuraminidasa H1N1
La búsqueda de la estructura primaria en NCBI de la H1N1 reportó una proteína humana compuesta por 454 aminoácidos y con peso molecular de 50,14 KDa (código NP_040981) (26). La composición de aminoácidos arrojada por el programa ProtParam (27) indicó que la serina (11,2%), la glicina (9,7%) y la isoleucina (9,0%) eran los aminoácidos más abundantes, mientras que la metionina (1,5%), la histidina (2,2%), la glutamina (2,4%) y la alanina (3,5%) estaban en baja proporción.
Por su parte, la búsqueda por homología en la PDB (algoritmo Blast) reportó una sola neuraminidasa de tipo N1, que coincidió con la H5N1 considerada en este estudio (PDB: 2HTY). El alineamiento de las secuencias procedentes de H1N1 y H5N1 mostró un porcentaje de identidad del 88%, valor que muestra la alta conservación de la estructura primaria en ambas proteínas, así como la utilidad del método para la predicción de estructuras terciarias. Además, al comprar las secuencias de ambas proteínas en torno al sitio activo se encontraron 11 aminoácidos altamente conservados (Figura 2).

Figura 2 Alineamiento secuencial entre H1N1 y H5N1. Los aminoácidos conservados se muestran en color rojo.
La predicción de la estructura secundaria de H1N1 mostró una composición del 6,83% en hélices alfa; 36,56% en hojas betas y 56, 61% en estructuras irregulares, evento que hace inferir una estructura poco definida. Estos resultados concuerdan con la composición porcentual de aminoácidos, en que los residuos involucrados en la formación de hélices alfa están en baja proporción (Met, His, Ala y Gln) mientras que los participantes en giros como la glicina están en alta proporción. Por su parte, el diagrama de Ramachandran (Figura 3) corrobora esta información al indicar la presencia de aminoácidos en regiones permitidas para hélices alfa y hojas betas.
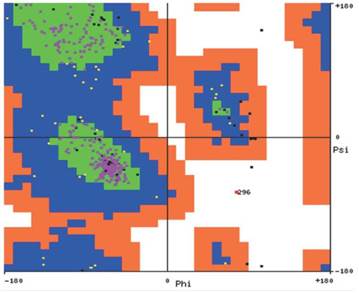
Figura 3 Diagrama de Ramachandran para la secuencia de H1N1. Se observa que la estructura secundaria de esta proteína contiene básicamente hélices alfa y hojas beta, las cuales pueden incluir giros.
En aras de obtener la estructura 3D de H1N1 se utilizó como plantilla la cadena A de la H5N1 (PDB: 2HTY) y los servidores de modelado por homología Swiss Model, Geno 3D y 3D-Jigsaw, los modelos obtenidos se muestran en la Figura 4.
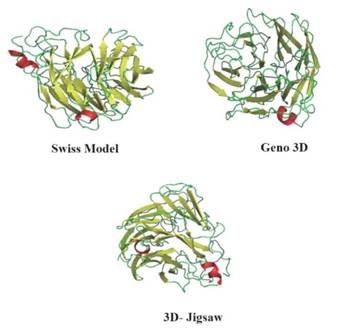
Figura 4 Modelos obtenidos por diversos servidores de modelado por homología. En cada caso se muestra la cadena principal y las cadenas laterales, hojas beta (amanillo), hélices alfa (rojo), loops (verde).
Como se muestra en la Figura 4, los tres modelos obtenidos evidencian una pequeña porción de hélices alfa en comparación a las hojas beta, así como una gran proporción de estructuras secundarias aleatorias como era de esperarse. El análisis de identidad de los tres modelos fue superior al 80%, por tanto, cualquiera de los tres modelos es viable para ensayos de acoplamiento molecular. No obstante, con el propósito de encontrar la mejor geometría se empleó como criterio la presencia de puentes disulfuro conservados en la estructura, así solo el modelo generado por Swiss Model mantuvo dichas interacciones igual al molde.
Por otro lado, el RMSD (Tabla 2) de los modelos obtenidos deja ver que 3D-Jigsaw presenta un valor óptimo, sin embargo, la energía es muy alta, lo que indica que no se favorece la estabilidad de la estructura. Por tanto, el modelo de Swiss continúa siendo la mejor opción debido a que tiene una mejor geometría y estabilidad con respecto a los otros modelos.
Tabla 2 Cálculo de RMSD y energía para los modelos obtenidos.
| Modelo | Molde | RMSD(Â) | Energía (KJ/ mol) |
|---|---|---|---|
| Swissmodel.pdb | 2HTY | 0,07 | -14.633,341 |
| geno.pdb | 2HTY | 1,13 | -13.067,107 |
| 3djigsaw.pdb | 2HTY | 0,25 | 6.274,353 |
La evaluación del modelo a través de los potenciales evidenció que Swiss Model, en tres de los cuatro programas utilizados (Eval23D, Verify3D y EvTree), mostró tener una estructura 3D óptima (Tabla 3).
Tabla 3 Evaluación del Swiss Model empleando los programas Eval23D, Verify3D, Prosa II y EvTree.
| PDB | Eval23D | Verify3D | Prosa II | EvTree |
|---|---|---|---|---|
| Swissmodel.pdb | 0,091 | 0,305 | - | 0,203 |
Los valores de evaluación van de -1 a 1 y son ilustrados con una franja de colores que va desde el rojo hasta el blanco para geometrías no favorables y desde el blanco hasta el verde para geometrías óptimas.
Tabla 4 Porcentajes de los residuos en las distintas regiones para el molde y el modelo.
| Molécula | Residuos en núcleo y permitida | Residuos en núcleo | Residuos en la región permitida | Residuos en la región generosa | Residuos en la región no permitida |
|---|---|---|---|---|---|
| Swissmodel.pdb | 100,0% | 72,5% (235) | 27,5% (89) | 0,0% (0) | 0,0% (0) |
| 2HTY | 98,5% | 79,9% (258) | 18,6% (60) | 1,2% (4) | 0,3% (1) |
La evaluación de calidad por estereoquímica indicó también la estabilidad tanto del modelo como del molde (2HTY) (Tabla 4). Los valores mayores al 90% indican las regiones favorecidas del diagrama de Ramachandran, considerando la estereoquímica Phi-Psi. Además, la tabla 3 muestra que en el Swiss Model no presentó ningún aminoácido con una conformación inconcebible, mientras que para el 2HTY solo un aminoácido adoptó dicha conformación, no obstante, este no hace parte del sitio activo ni es un residuo cercano. También, la calidad del modelo por normalidad (Z-score) mostró que la estructura generada por el Swiss Model y el molde están dentro de los Z-score de las proteínas nativas reportadas en la Protein Data Bank. Por tanto, todos los parámetros considerados en la evaluación del molde y el Swiss Model indicaron su estabilidad y geometría favorable para su utilización en los ensayos de acoplamiento molecular.
Acoplamiento molecular de ligandos - neuraminidasa H1N1
Con el propósito de explicar los resultados del acoplamiento molecular se considera la nomenclatura de los ligandos mostrada en la Figura 5.
Complejos neuraminidasa H1N1-F36 y H1N1-Ginkgetin
Al analizar el complejo H1N1-F36 se observa la formación de seis puentes de hidrógeno, los cuales están distribuidos entre el sitio activo (Arg278 y Tyr387) y el resto de la cavidad (Arg141y Asn329) (figura 6a). El ligando F36 forma dos puentes de hidrógeno con los grupos NH2 de Arg278, uno con el radical OCH3 en C8 y el otro con el radical OH en C7. El residuo Asn329 lo hace a través del grupo carbonilo y átomo de hidrogeno del radical OH en C7, quien a su vez hace otro puente con el grupo NH2 de este residuo, por tanto, la importancia del radical OH-C7 en la estabilización del complejo. Por su parte, el residuo Tyr387 del sitio activo forma un puente de hidrógeno con O1 del anillo de pirona de F36 mientras que Arg141 lo hace a través del grupo NH2 y el O del radical OH en C4'. También, el complejo H1N1-F36 se estabiliza con las interacciones hidrofóbicas entre el anillo aromático del ligando y los residuos de Arg103, Asp136 y Leu119.
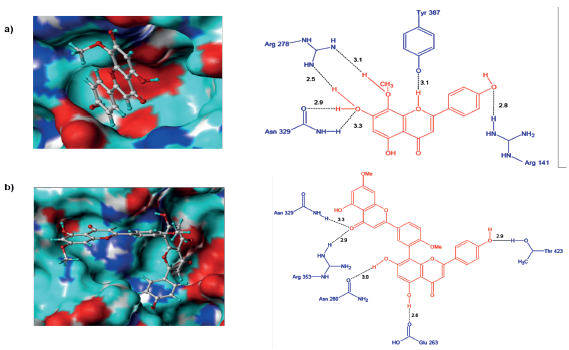
Figura 6 Conformación de los ligandos e interacciones entre el sitio activo de la neuraminidasa H1N1 y a) F36 y b) Ginkgetin.
En el caso de Ginkgetin adoptó una conformación poco favorable dentro del sitio activo de H1N1, debido a que solo una parte de este ingresó a la cavidad (Figura 6b). Este hecho podría entonces explicar su baja capacidad inhibitoria (Tabla 1). Al analizar las interacciones se observó que Ginkgetin formó cinco puentes de hidrógeno, siendo el más relevante el establecido entre Arg353 (sitio activo) y el grupo carbonilo de C4, así mismo, este grupo también interactuó con Asn329. Los demás puentes se formaron con Asn280, Glu263 y Thr423 y los grupos OH del ligando. Por otro lado, a diferencia del complejo H1N1-F36, este presenta solo una interacción de tipo hidrofóbico (Arg353), lo cual corrobora su baja estabilidad dentro del sitio activo.
Complejos neuraminidasa H1N1-3R y H1N1-3S
Al revisar la conformación del ligando 3R dentro de la proteína H1N1, se observó que su contacto principal fue a través del sustituyente de ácido siálico acetilado, quedando por fuera la parte biflavonoide; situación que es de esperarse debido al gran volumen de 3R (Figura 7). Las pocas interacciones de este complejo se establecieron mediante cuatro puentes de hidrógeno formados entre Arg278, Arg353 y el oxígeno de C7", Arg278, Tyr378 y el sustituyente ácido siálico acetilado, mientras que las interacciones hidrofóbicas se presentaron con los residuos Pro416, Thr423 y Glu262 como se muestra en la Figura 7.
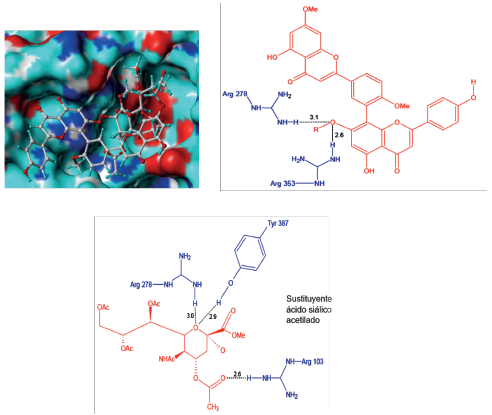
Figura 7 Conformación de los ligandos e interacciones entre el sitio activo de la neuraminidasa H1N1 y el ligando 3R.
Además, se encontraron interacciones de tipo repulsivas, lo cual corrobora su desfavorable acople e indica la posible razón de su baja actividad inhibitoria (Tabla 1). Por su parte, el ligando 3S no se acopló al sitio activo de H1N1, hecho que evidenció la baja estabilidad del complejo y por tanto su pésima actividad inhibitoria.
Complejos neuraminidasa H1N1-5R y HN1-5S
El ligando 5R, a diferencia de los mencionados, ingresa completamente al sitio activo de H1N1; situación que brinda una mayor estabilidad al complejo (Figura 8a). La interacción entre 5R y H1N1 se da a través de la formación de nueve puentes de hidrógeno de los cuales cuatro son con la parte biflavonoide y cinco con el sustituyente ácido siálico acetilado. La parte biflavonoide interacciona con dos residuos del sitio activo (Arg103 y Asp136) mientras que el ácido siálico acetilado con tres (Arg278, Arg353 y Tyr387), como se muestra en la Figura 8a. Las interacciones hidrofóbicas se presentaron entre el anillo de benceno del extremo superior del flavonoide y la Pro416, evento que favorece aún más la estabilidad del complejo, tanto a nivel estructural como energético. Todo lo anterior puede dar explicación a la alta actividad inhibitoria de esta molécula.
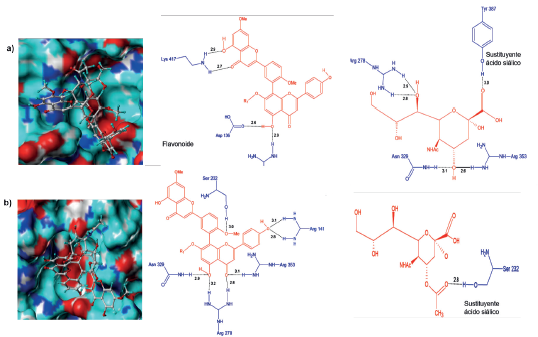
Figura 8 Conformación de los ligandos e interacciones entre el sitio activo de la neuraminidasa H1N1 y a) 5R y b) 5S.
Por su parte, el complejo 5S-H1N1 mostró que el ligando adoptó una conformación similar a la de 5R dentro de sitio activo de la proteína (Figura 8b). La parte biflavonoide formó cinco puentes de hidrógeno, siendo dos de ellos con residuos del sitio activo (Arg278 y Arg353), mientras que el sustituyente ácido siálico acetilado solo tuvo un contacto (Figura 8b). Los contactos hidrofóbicos se establecieron con los residuos Pro231, Lys135, Ile208, y Asp136, resaltando que los dos últimos pertenecen al sitio activo de la proteína. Por tanto, esta información deja en evidencia que la actividad inhibitoria de los ligandos 5R y 5S está relacionada con su capacidad de acople al sitio activo y el tamaño de sus sustituyentes.
Complejos neuraminidasa H1N1-65R y HN1-6S
El complejo H1N1-6R se muestra en la Figura 9a. Al igual que los ligandos de la serie cinco, 6R se ubicó de forma favorable dentro del sitio activo gracias a la formación de puentes de hidrógeno e interacciones hidrofóbicas. Es así como la parte flavonoide de 6R formó tres puentes de hidrógeno con los residuos Arg103 (sitio activo) y Thr423, como se observa en la Figura 9a. Por su parte, el sustituyente ácido siálico formó seis puentes de hidrógeno con cinco residuos, siendo cuatro de ellos del sitio activo (Arg103, Arg353, Arg278, Tyr387) y la Glu263 (figura 9a).
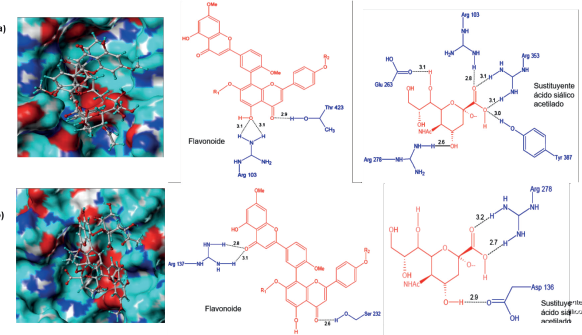
Figura 9 Conformación de los ligandos e interacciones entre el sitio activo de la neuraminidasa H1N1 y a) 6R y b) 6S.
En general 6R estableció nueve puentes de hidrógeno, de los cuales cuatro pertenecen al sitio activo, en el que se destacan los formados con Arg353 y Arg103. Los contactos hidrofóbicos más importantes se formaron con Pro416, Thr423 y Asp136. La Pro416 interactúa con el radical metilo y todo el anillo de benceno superior del biflavonoide, mientras que Thr423 lo hace con los dos anillos de benceno adyacentes al biflavonoide (3' - 8"). Por otro lado, el complejo H1N1-6S al igual que H1N1-6R mostró ser energéticamente favorable (Figura 9b). La parte biflavonoide formó tres puentes de hidrógeno, uno con Ser232 y dos con Arg137 (sitio activo). En lo que respecta al ácido siálico, se observó la formación de tres puentes de hidrógeno dos con Arg278 y uno con Asp136; ambos residuos hacen parte del sitio activo de H1N1.
Con base en la información descrita, se pudo establecer que la actividad inhibitoria de los ligandos con H1N1 está mediada por la formación de interacciones tipo puente de hidrógeno e hidrofóbicas, es así como se encontró que 3S tiene pocas interacciones, hecho consecuente con baja actividad inhibitoria (IC50 > 100), mientras que el ligando 5R (IC50 = 5,50), con la actividad más alta mostró un mayor número de interacciones. También, el acople de los ligandos al sitio activo de la proteína indicó que estos actúan como inhibidores competitivos y que la estereoquímica R del C2 en el sustituyente ácido siálico favorece a una mejor interacción con la proteína, debido a su menor volumen y la presencia de los grupos hidrofílicos OH.
Acoplamiento molecular complejo ligandos-neuraminidasa H5N1
Complejos neuraminidasa H5N1-F36 y H5N1-Ginkgetin
El complejo F36-H5N1 muestra que el ligando F36 se acopla de manera favorable al sitio activo de la proteína a través de la formación de puentes de hidrógeno y contactos hidrofóbicos (figura 10a). Los siete puentes de hidrógeno se presentaron con cinco residuos de aminoácidos, de los cuales solo la Tyr347 no pertenece al sitio activo, como se observa en la figura 10a. Los residuos Arg118 y Arg371 con sus grupos NH2 de las cadenas laterales interaccionan fuertemente formando cuatro enlaces con OH de C5 y OCH3 de C8 respectivamente, mientras que el residuo Tyr347 forma un enlace con O1 del anillo de pirona. El OH de Tyr406 y el carbonilo de Asp151 hacen lo propio con los sustituyentes OH de C4' y C5, respectivamente. Los contactos hidrofóbicos presentes en este complejo los constituyen las partes apolares de los aminoácidos citados con los anillos aromáticos del ligando. Un análisis comparativo entre los complejos del ligando F36 con las neuraminidasas H1N1 y H5N1 muestra una tendencia a conservar las interacciones por puentes de hidrógeno en su mayoría con residuos del sitio activo, siendo los puntos críticos el sustituyente OH de C4', el radical OCH3 de C8 y el O del anillo de pirona, así como los anillos de benceno que participan en los contactos hidrofóbicos.
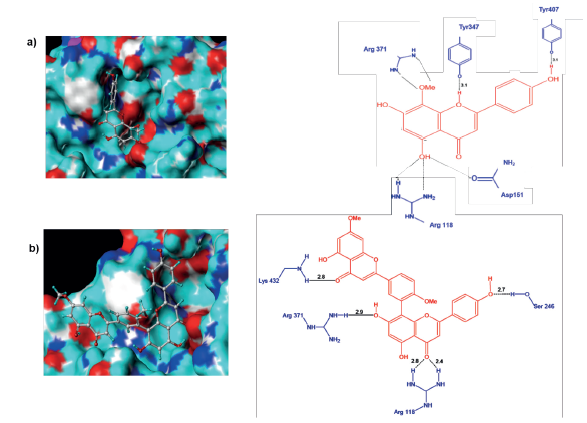
Figura 10 Conformación de los ligandos e interacciones entre el sitio activo de la neuraminidasa H5N1 y a) F36 y b) Ginkgetin.
Para el caso de Ginkgetin (figura 10b), el complejo evidenció que solo la parte flavonoide interactúa con el sitio activo de la proteína, comportamiento que es similar al observado con H1N1. En general, la Ginkgetin formó cinco puentes de hidrógeno con cuatro aminoácidos, de los cuales dos corresponden al sitio catalítico (Arg118 y Arg371) (figura 10b). La Arg118 se enlaza fuertemente con sus NH2 formando dos enlaces con el carbonilo de C4" del anillo de pirona, al igual que Lys432 que se une al otro anillo de pirona con el oxígeno carbonilo de C4. Los sustituyentes OH de C7" y C4'" forman puentes de hidrógeno respectivamente con Arg371 y Ser246. Los puntos críticos de estabilización de Ginkgetin tanto en H1N1 y H5N1 corresponden a los grupos OH de C4'" y C7", así como el oxígeno carbonilo de C4. Además, el complejo con H5N1 se estabiliza más por contactos Fhidrofóbicos que el correspondiente complejo con H1N1 (figuras 11a y 11b). No obstante, los complejos formados por Ginkgetin en ambos casos son inestables, hecho que desfavorece su actividad inhibitoria.
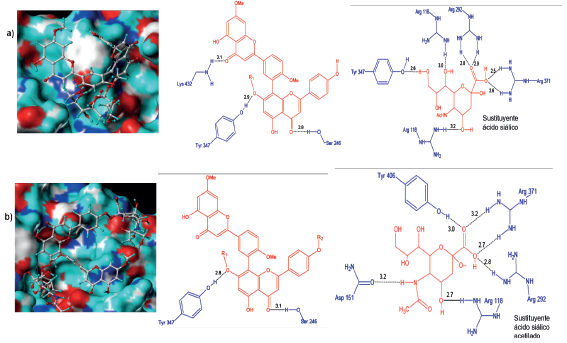
Complejos neuraminidasa H5N1-5R y H5N1- 6R
El complejo formado por 5R al igual que su homólogo H1N1 se acopló energéticamente favorable con el sitio activo (Figura 11a). La parte del biflavonoide de 5R formó puentes de hidrógeno con los residuos Lys432, Tyr347 y Ser246, los cuales no pertenecen al sitio activo de la proteína, sin embargo, Lys432 y Ser246 toma relevancia puesto que participaron en la estabilización de los complejos formados con F36 y Ginkgetin. La parte del ácido siálico desempeña un papel importante en el acople de la molécula, debido a que ella interactúa de forma mayoritaria con el sitio activo de H5N1. Es así como siete puentes de hidrógeno se establecieron con los residuos Arg118, Arg292, Arg371 (Sitio activo) y Tyr347. Las Arg371 y Arg292 con sus grupos NH2 ataca al grupo carboxilo en C1, por tanto, este radical se convierte en un punto crítico del ligando, mientras que Arg118 forma dos puentes de hidrógeno con los grupos OH en C4 y C6, respectivamente, y Tyr347 con el radical OH en C9. En cuanto a los contactos hidrofóbicos se encontró que el residuo Pro431 interacciona fuertemente con la cadena carbonada del extremo del ácido siálico y el anillo de la Tyr347 con el metilo del grupo OCH3 ubicado en C7.
Por otro lado, el ligando 6R a diferencia de 5R se acomoda en el sitio activo de H5N1 a través del sustituyente de ácido siálico de la posición R1 y una pequeña porción del biflavonoide (figura 11b). El análisis de interacciones mostró que el sustituyente ácido siálico de R1 estableció seis puentes de hidrógeno con la cavidad (Try406, Arg371, Arg292, Arg118, Asp151), mientras que dos con la parte flavonoide (Tyr347 y Ser 246). Los contactos hidrofóbicos fueron mediados por la cadena apolar del ácido siálico y Asp 198, Ile222 y Asn221. La suma de las interacciones evidencia la estabilidad del complejo mostrado en la figura 11b.
Conclusiones
Este estudio permitió obtener la estructura tridimensional de la neuraminidasa H1N1 mediante el modelado por homología, usando como plantilla la proteína H5N1 disponible en el Protein Data Bank (2HTY). De acuerdo con los parámetros evaluados, este modelo mostró ser geométrica y energéticamente estable, convirtiéndose en material disponible para ensayos in silico que requieran de esta estructura. El análisis del acoplamiento molecular permitió evidenciar la relación entre el poder inhibitorio de los análogos de flavonoides derivados de amentoflavona y las interacciones establecidas con el sitio activo de las neuraminidasas H1N1 y H5N1 del virus de la gripa aviar. Es así como los ligandos 5R y 6R con las mejores actividades inhibitorias contra H1N1 mostraron tener un mayor número de interacciones del tipo puente de hidrógeno e hidrofóbicas con el sitio activo de ambas proteínas. Además, se resalta el papel de la estereoquímica R sobre la S, debido a que la primera ofrece más flexibilidad al ligando y, por ende, brinda el espacio para formar un número mayor de interacciones. Por otro lado, los ligandos 5R y 6R acoplados con H1N1 mostraron que la actividad inhibitoria mejora cuando el grupo en R1 tiene poco volumen (5R), ya que las interacciones se distribuyen de forma similar entre la parte biflavonoide y el sustituyente de R1, lo cual confiere una mayor estabilidad al complejo. Por tanto, este trabajo propone la evaluación experimental de los ligandos 5R y 6R como potenciales inhibidores de H5N1 y posterior uso en ensayos clínicos.

