











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
La ecología química es un campo de conocimiento transdisciplinar que cada vez gana mayor relevancia [1], [2]. Parte fundamental de esta disciplina es la identificación de las moléculas que median la comunicación entre insectos o planta-insecto. En la medida en que se conocen más semioquímicos y se acumulan datos sobre su uso, resulta conveniente la creación de herramientas que contribuyan de una manera rápida y eficiente a establecer patrones y relaciones sobre esta información y así aportar a futuras investigaciones.
En este como en otros campos de la química, las relaciones entre estructura y actividad están en el centro del problema y parte fundamental de este es lograr una buena representación cuantitativa de la estructura química; es decir, una que pueda procesarse computacionalmente [3]. De ahí que actualmente se hayan definido miles de descriptores moleculares que buscan codificar distintos aspectos de la estructura; por ejemplo, características topológicas, propiedades fisicoquímicas, grupos funcionales u otras propiedades dependientes de la conformación espacial de los componentes moleculares [4]. Sin embargo, la selección de los descriptores apropiados es un problema abierto que depende del objetivo que se persigue [5], [6]. Alternativamente, se puede acudir a conjuntos redundantes de los mismos como estrategia multiobjetivo para la búsqueda de patrones [7], [8].
El sistema insecto-semioquímico y la representación de estas moléculas constituyen un sistema complejo para el cual se ha generado y se sigue generando información. Este sistema constituye un espacio propicio para ser explorado mediante técnicas de minería de datos y aprendizaje de máquina, cuya utilización en diversos campos de la ciencia y la tecnología ha sido exitosa. En química, por ejemplo, se han implementado algoritmos de aprendizaje automático de distinta naturaleza que solventan problemas en diversas áreas, que abarcan desde la química analítica hasta la catálisis, pasando por la química orgánica y la química computacional [9]. Algunos de estos estudios apuntan a tratar relaciones estructura-actividad (SAR) por medio del desarrollo de herramientas de aprendizaje automático que facilitan el diseño y la selección de moléculas con la actividad esperada. En particular, algunos hacen uso de perceptrones multicapa y redes neuronales profundas para la clasificación de moléculas [10], [11].
En este trabajo nos proponemos comparar un conjunto de metodologías para la selección de las variables más relevantes y métodos de agrupamiento que pueden ser empleados para el descubrimiento de patrones estructurales en el conjunto de los semioquímicos reportados para los coleópteros de la familia Scarabaeidae. A la vez, implementamos modelos de aprendizaje de máquina capaces de realizar la clasificación de este tipo de metabolitos en las categorías establecidas por el patrón descubierto.
Materiales y métodos
Sistema de estudio
Para este estudio se empleó un modelo constituido por un conjunto de 148 semioquímicos reportados para 240 especies de insectos de la familia Scarabaeidae (orden: Coleóptera) que se almacenó en una base de datos relacional SQL [12]1.
La estructura química de los compuestos considerados se caracterizó mediante un conjunto redundante de descriptores moleculares de diferentes clases. Así, con el programa RDKit [13] se calcularon 188 descriptores, los cuales clasificamos en cuatro clases: la primera constituida por descriptores 19 derivados esencialmente de propiedades grafo-teóricas, la segunda corresponde a 106 descriptores que dan cuenta de la constitución de las moléculas según la presencia de ciertos fragmentos o tipos de enlaces, la tercera consta de 58 descriptores basados en propiedades que se calculan sobre superficies tridimensionales asociadas a las moléculas y la cuarta está conformada por cinco descriptores asociados a propiedades fisicoquímicas. El listado de los descriptores calculados puede consultarse en la siguiente dirección URL: https://www.rdkit.org/docs/GettingStartedInPython.html#descriptor-calculation.
Además, se calcularon 12 descriptores de naturaleza cuántica (HOMO; LUMO; constantes rotacionales en X, Y y Z; extensión espacial electrónica; momento dipolar en X, Y, Z y el total; electronegatividad y dureza) derivados de la matriz de densidad obtenida con el método de funcionales de la densidad B3LYP/6-31(d,p) implementado en Gaussian 09 [14]. De esta forma se propone un total de 200 descriptores moleculares de cinco clases diferentes.
Dado que hemos seleccionado un conjunto redundante de descriptores, es posible que se presenten dependencias entre algunos de ellos o que hayamos incluido algunos poco relevantes en el momento de reproducir un patrón de clasificación; por lo tanto, es necesario seleccionar los descriptores más discriminatorios para evitar sesgos. Este proceso se llevó a cabo de manera paralela mediante tres métodos:
(i) Análisis de componentes principales (ACP) [15]: el ACP suele emplearse para reducir la dimensionalidad del espacio de representación; para ello se definen unas nuevas variables mediante una transformación que asegura que la mayor parte de la varianza de los datos sea explicada por unas pocas de estas variables, los componentes principales. Cuando se emplea para seleccionar entre las variables originales deben escogerse aquellas (los descriptores) que más contribuyen a la conformación de los componentes principales; como criterio de selección se propuso que la suma de los cuadrados de los coeficientes del descriptor con que contribuye a los componentes principales, que describen hasta el 70% de la varianza acumulada de los datos, tuviese un valor superior a 0,3 (la suma sobre todos los componentes principales es por definición la unidad). La selección de descriptores se realizó por separado para las cinco clases mencionadas.
(ii) Bosques aleatorios (BA) [16]: BA es un método de aprendizaje supervisado, basado en árboles de decisión, para clasificar objetos en función de un patrón previamente determinado. Este método establece unas puntuaciones de importancia para cada variable según la impureza de Gini, la cual es una medida que evalúa la distribución de los datos por nodo y establece qué tan óptima es una escisión de los datos respecto a cada variable. Estas puntuaciones de importancia permiten seleccionar las variables más significativas para reproducir una clasificación propuesta; para este estudio se seleccionaron los descriptores con puntuaciones superiores a 0,41%; este valor nos permitió tener el mismo número de variables que fue hallado mediante el uso de ACP.
(iii) Boruta-Shap (BS) [17]: BS es un método de selección de variables que combina el algoritmo Boruta y la técnica Shap. El primero realiza una selección y eliminación iterativa de variables no relevantes para una función objetivo o patrón de clasificación, teniendo como criterio las puntuaciones de importancia de unas variables "sombra" (las cuales son una combinación aleatoria de las variables originales); y la segunda establece las variables que presentan una mayor influencia en las predicciones de los modelos de aprendizaje de máquina con base en la teoría de juegos cooperativos.
Para el ACP se empleó la librería stasts disponible para lenguaje de programación R [18] y para los métodos de BA y BS se emplearon las librerías SciKit-learn [19] y Boruta-Shap [17], respectivamente disponibles en Python2. Los tres resultados de selección de variables se comparan más adelante.
Reconocimiento de patrones y técnicas de agrupamiento
Si bien existe una gran cantidad de métodos para el descubrimiento de patrones que utilizan algoritmos no supervisados, en este trabajo se utilizaron dos técnicas diferentes: el método de lógica difusa C-means (FCM, por su nombre en inglés) [20] y los mapas autoorganizados de Kohonen (SOM, por sus siglas en inglés) [21]. Estos últimos con la intención de corroborar la plausibilidad de posible patrón hallado con el primero.
Los métodos de agrupamientos no jerárquicos como el FCM requieren de la definición a priori de un número de centroides o semillas. Probamos un número de semillas entre 2 y 5 y que en todos los casos se observan tres grupos; alternativamente ensayamos diversos métodos de agrupamiento jerárquico, pero no logramos la reproducibilidad de ninguna clasificación [22]. El algoritmo FCM se ejecutó en R [18] con un valor de 2 para el parámetro de difuminado y un criterio de convergencia de 1*10-9.
Las redes neuronales SOM fueron construidas en Python [19] usando la biblioteca MiniSOM [23] y constan de m neuronas de entrada; m corresponde al número de variables seleccionadas (descriptores moleculares relevantes) que alimentan la red, y 225 neuronas de salida (15x15); lo cual corresponde a la arquitectura óptima respecto al error de cuantización y al número de neuronas que han de especializarse en cada variable. Las SOM fueron entrenadas con una tasa de aprendizaje de 0,01, con un factor sigma de 1,5, durante 1.000 épocas, y como función de vecindad se empleó una función gaussiana. La arquitectura de este sistema neuronal corresponde a una red cuadrada. Para su entrenamiento se emplearon los tres conjuntos de descriptores determinados como los más significativos con los tres métodos mencionados anteriormente [21].
Para clasificar las moléculas no adscritas con certeza a alguno de los grupos encontrados se diseñó y construyó un clasificador multiclase, un perceptrón multicapa (MLP) [24]. Se trata de un algoritmo basado en aprendizaje supervisado, conformado por cinco capas de neuronas: una de entrada de 53 neuronas, una de salida de tres neuronas y tres capas ocultas con 60, 40 y 20 neuronas, respectivamente. Para esta red neuronal se utilizó la función sigmoidea como función de activación y el optimizador de costos "adam" con una tasa de aprendizaje de 0,0001. Esta red neuronal también fue programada en Python usando la biblioteca SciKit-learn [19].
Resultados y discusión
Para consignar la información en nuestra base de datos, se partió de la reportada, para el suborden Polyphaga, en Pherobase [25]. Esta última es una base de datos de libre acceso tanto para consulta como para el registro de datos, por lo cual es común que presente inconsistencias e información errada3. En consecuencia, se depuró la información consultando la bibliografía primaria, es decir, un total de 957 artículos reportados en la literatura especializada.
Como anotamos, para caracterizar la estructura química de los semioquímicos empleamos un conjunto redundante de descriptores; por lo tanto, la información que codifican algunas de estas variables puede referirse a un mismo aspecto, ya sea porque fueron propuestos para dar cuenta de una misma característica estructural o bien porque fueron derivados de un mismo descriptor fundamental por autores diferentes. Consideramos que usar un conjunto redundante de variables se justifica en la medida en que no existe el fundamento teórico que permita caracterizar de una manera cuantitativa y unívoca el concepto de estructura química, quizá uno de los más complejos de esta ciencia. La selección de los descriptores más relevantes la hicimos primero mediante ACP y, como mencionamos, la selección de las variables originales que más participan en los componentes principales se realizó por separado para cada una de las seis clases de descriptores moleculares. Este procedimiento lo planteamos con el fin de asegurar que el conjunto final mantuviese lo esencial que pretende codificar cada una de las seis clases de descriptores. Por último, constatamos que no existe mayor correlación entre las variables seleccionadas. Esta metodología nos permitió reducir el conjunto de descriptores; así, pasamos de un conjunto de 200 a uno de 53, que reconocemos como las variables más significativas.
Los semioquímicos considerados fueron agrupados mediante el método FCM; como anotamos anteriormente, se observa la existencia de tres grandes grupos. Al comparar los valores de pertenencia de cada molécula a cada grupo notamos que para el grupo R1 hay moléculas con pertenencia superior a 0,85. Para los grupos R2 y R3 las pertenencias más altas apenas superan un valor de 0,45; estos valores indican que hay moléculas con pertenencias similares a estos dos grupos. Escogimos las moléculas con mayor pertenencia a cada grupo para definir su núcleo, de manera que pudiésemos centrar nuestra atención en pequeños conjuntos de moléculas que según FCM deberían ser bastante similares entre sí. Así fue posible identificar un patrón estructural: el núcleo del grupo R1 está formado por hidrocarburos lineales; el núcleo del grupo R2 incluye moléculas aromáticas o con dobles enlaces conjugados y el núcleo del grupo R3 por moléculas de bajo peso molecular que incluye alcoholes, aldehídos, cetonas y ésteres. (Ver Tabla 1).
Tabla 1 Ejemplo de moléculas pertenecientes al núcleo de cada grupo, los valores de pertenencia y su asociación con su respectiva ruta biosintética [26].
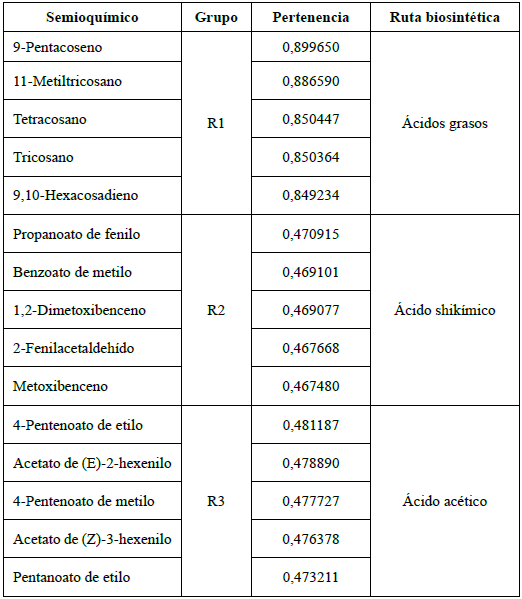
Vale la pena destacar que las moléculas que conforman cada uno de los tres núcleos propuestos corresponden a metabolitos secundarios que comparten un mismo origen biosintético; por lo tanto, el patrón estructural que se develó con FCM corresponde a la clasificación según las respectivas rutas biosintéticas [26], al menos para las moléculas de alta pertenencia a cada grupo (Véase Figura 1).

Figura 1 Representación del grado de pertenencia de cada una de las moléculas a los tres grupos establecidos por el método de lógica difusa FCM. Para el grupo R1, se observa una pertenencia superior a 0,8 para un grupo pequeño de moléculas. Para los otros dos grupos, R2 y R3, las moléculas tienen grados de pertenencia bastante parecidos, aunque bajos; las de mayor pertenencia se diferencian en milésimas.
Una vez hemos reconocido un patrón correspondiente a las rutas biosintéticas, podemos emplearlo como la función de respuesta que requieren las técnicas de BA y BS para hacer la selección de descriptores. Para la técnica de bosques aleatorios, al igual que en el ACP, obtuvimos un conjunto final de 53 descriptores y con Boruta-Shap este conjunto aumentó a 60 variables4. Al comparar los tres conjuntos de variables seleccionadas, encontramos que los métodos de selección dirigida, los cuales consideran el conjunto total de variables, dan lugar a conjuntos de variables óptimas similares a los que hallamos mediante ACP, que realizamos por aparte para cada clase de descriptores. El número de descriptores por tipo seleccionados con cada técnica puede verse en la Tabla 2.
Tabla 2 Distribución de descriptores moleculares seleccionados mediante cada metodología por clase de descriptor.
| Técnicas de selección de características | |||
|---|---|---|---|
| Clase de descriptores | ACP | BA | BS |
| Calculados sobre superficies (3D) | 11 | 14 | 18 |
| Constitucionales | 20 | 12 | 12 |
| Cuánticos | 10 | 5 | 8 |
| Grafo-teóricos | 10 | 18 | 18 |
| Propiedades fisicoquímicas | 2 | 4 | 5 |
Por otra parte, al repetir el agrupamiento con el FCM con las variables derivadas de BA y BS encontramos que los núcleos en cada agrupamiento se conservan. Para corroborar estos agrupamientos empleamos mapas autoorganizados de Kohonen (SOM). El entrenamiento y la evaluación de los SOM se llevó a cabo con los 102 compuestos que se encuentran en los núcleos de los grupos derivados del FCM y los tres conjuntos de variables.
Como resultado obtuvimos tres redes neuronales que logran reproducir la clasificación previamente conseguida según la ruta biosintética, como se muestra en la Figura 2. En ella se observa que el mejor agrupamiento se obtuvo al emplear los descriptores derivados del método de bosques aleatorios, pues separa de mejor manera las distintas moléculas por fronteras de neuronas con baja activación (ver Figura 2), y además presenta el error de cuantización [21] más bajo de las tres: 1,68. A su vez, los SOM alimentados con los descriptores derivados del ACP y BS no logran separar tan claramente algunas moléculas; este es el caso de los subgrupos que se forman para la ruta del ácido shikímico (R2) y la ruta del ácido acético (R3), según se aprecia en las Figuras 2a y 2c; además, el error de cuantización fue mayor: 2,78 y 2,04, respectivamente.
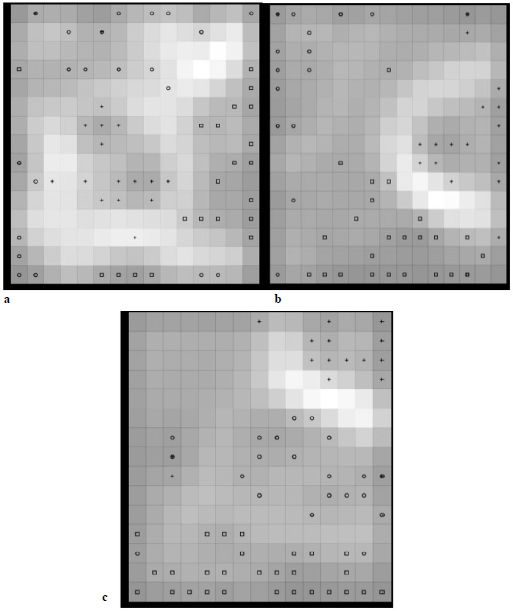
Figura 2 Mapas autoorganizados de Kohonen entrenados con los descriptores resultantes de la selección por: a. Análisis de componentes principales, b. Bosques aleatorios y c. Boruta-Shap. Las neuronas que presentan un color más claro tienen una menor activación y constituyen las fronteras entre los grupos: O Ruta del ácido shikímico, + Ruta de los ácidos grasos, + Ruta del ácido acético. Nótese que algunas moléculas pertenecientes a rutas distintas activan una misma neurona.
Si bien fue posible hallar un patrón a partir del método de agrupamiento difuso, y se corroboró la consistencia del mismo con los SOM, aún persisten 46 moléculas que no presentan un grado claro de pertenencia a alguna agrupación o ruta biosintética. Para complementar el modelo de aprendizaje de máquina que estamos presentando, empleamos perceptrones multicapa para clasificar estos 46 semioquímicos. Presentamos dos MLP, uno que usa el conjunto de descriptores determinado por ACP (MLP//ACP), pues fue el punto de partida para establecer el patrón de clasificación, y el otro con los descriptores seleccionados por el método de BA (MLP//BA), ya que estos demostraron ser el conjunto de variables más apropiado para diferenciar las clases en que buscamos clasificar los semioquímicos. Al igual que los SOM, los MLP fueron entrenados con los mismos 102 compuestos bien definidos. Este conjunto se dividió aleatoriamente en una proporción 80-20, en que el 80% de los semioquímicos se empleó para el entrenamiento y el 20% restante se utilizó como conjunto de prueba.
Para ambos modelos se obtuvo una concordancia del 100% entre la respuesta obtenida y la respuesta esperada para el conjunto de entrenamiento. En cuanto al conjunto de prueba, el modelo MLP//ACP presentó una concordancia del 95%, mientras que para el modelo MLP//BA la concordancia fue del 100%. La precisión y la sensibilidad en la clasificación del conjunto de prueba se pueden ver en la Tabla 3. Estos dos modelos se emplearon para establecer las rutas biosintéticas del conjunto de 46 moléculas problema. Dado que se trata de un conjunto pequeño de compuestos a clasificar, pudimos revisar de manera manual la predicción para cada uno de ellos. Así, con los dos modelos 31 de las 46 moléculas son clasificadas de la siguiente manera: 18 en la ruta del ácido shikímico (R2), nueve en la ruta del ácido acético (R3) y cuatro en la ruta de los ácidos grasos (R1). No obstante, de estas 31 moléculas ocho no parecen bien clasificadas; se trata de terpenoides que en principio deberían estar en la ruta R3, pero que se reparten entre la R1 y la R2. Consideramos que esto puede deberse a que la biosíntesis de esta clase de metabolitos involucra múltiples mecanismos que generan una gran diversidad estructural [27] o bien a una deficiente representación de este tipo de compuestos. No obstante, quizá lo más factible es que estas clasificaciones erradas se deban al tamaño de la muestra empleada, en la cual no se cuenta con suficiente representación de todas las posibles categorías.
Tabla 3 Precisión y sensibilidad para las clasificaciones obtenidas para el conjunto de prueba con cada modelo.
| Rutas biosintéticas | Precisión | Sensibilidad | ||
|---|---|---|---|---|
| MLP//ACP | MLP//BA | MLP//ACP | MLP//BA | |
| R2 | 1,00 | 1,00 | 0,86 | 1,00 |
| R3 | 0,88 | 1,00 | 1,00 | 1,00 |
| R1 | 1,00 | 1,00 | 1,00 | 1,00 |
Por otra parte, quedan 15 moléculas para las cuales se predicen clasificaciones distintas con cada modelo. Al respecto, debe tenerse en cuenta que algunas moléculas se pueden obtener tanto de la ruta de los ácidos grasos como de la ruta del ácido acético; esto podría explicar lo que ocurre con seis de las 15 moléculas (ver Tabla 4, aquellas marcadas con *). Además, debemos recordar que la ruta del ácido acético también da origen a compuestos aromáticos, lo que podría justificar la clasificación de tres de las nueve restantes (ver Tabla 4, aquellas marcadas con f), resultado que solo se presenta para el MLP//ACP. Las otras seis están bien clasificadas por alguno de los dos modelos.
Tabla 4 Moléculas con un grado de pertenencia no definido en FCM y para las cuales se presentan resultados ambivalentes empleando los descriptores derivados del ACP y determinados por bosques aleatorios usando el perceptrón multicapa.
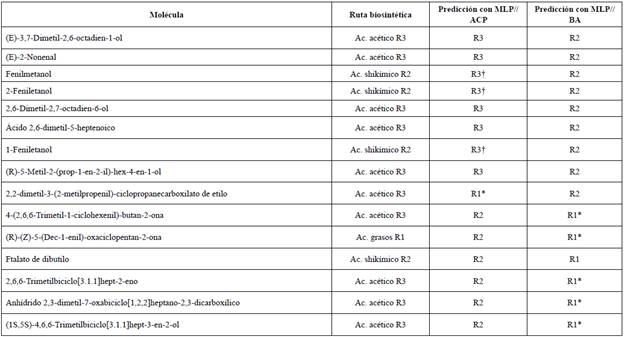
En vista de que algunas equivocaciones en la clasificación son más graves que otras, empleamos el coeficiente Kappa de Cohen ponderado [28] para comparar los modelos y evaluar el nivel de acuerdo entre las predicciones y las clasificaciones. Para ello propusimos las siguientes penalidades: tres para las moléculas que presuntamente derivan del ácido shikímico y los perceptrones las clasifican como compuestos derivados de la ruta del ácido acético o de los ácidos grasos y uno para los otros dos casos. Así, Kappa para el perceptrón entrenado con los descriptores derivados de bosques aleatorios (MLP//BA) es de 0,57, mientras que para el modelo entrenado con los descriptores derivados del ACP (MLP//ACP) es de 0,54. En ese sentido, se presenta un mayor acuerdo entre la respuesta esperada y su predicción para el modelo MLP//BA. Si bien el valor к = 0,57 se interpreta como un resultado aceptable para las clasificaciones, ha de tenerse en cuenta que el sistema aquí considerado cuenta con muy pocos registros y, a pesar de esto, la metodología propuesta arroja resultados promisorios.
Conclusiones
Conseguimos ensamblar un conjunto de técnicas de aprendizaje de máquina que conforman un modelo para el descubrimiento de patrones sobre conjuntos de compuestos y la posterior clasificación de moléculas problema en función del patrón establecido.
Los descriptores seleccionados a partir del análisis de componentes principales en combinación con la técnica de agrupamiento difuso C-means nos permitió reconocer un patrón entre los semioquímicos estudiados, el cual corresponde a las rutas biosintéticas que dan lugar a estos metabolitos secundarios.
Una vez identificado un patrón, recomendamos la técnica de bosques aleatorios para seleccionar las variables. Finalmente, la combinación de la metodología propuesta con un perceptrón multicapa nos permitió alcanzar una asignación aceptable de las rutas biosintéticas para las moléculas que C-means no clasificó.
Las moléculas cuya clasificación no fue del todo satisfactoria nos muestran la necesidad de ampliar el conjunto de información, para incluir una mayor variabilidad estructural que lleve a conjuntos de entrenamiento más robustos.

