










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Agronómica
Print version ISSN 0120-2812
Acta Agron. vol.58 no.1 Palmira Jan./March 2009
RESUMEN
La población S1 de la variedad tailandesa de yuca MTAi8 (AM320), la cual presenta patrones de segregación definidos para el contenido de carotenos totales (Beta-caroteno), se sometió a un análisis de agrupamiento segregante (BSA = Bulk Segregant Analysis) empleando 700 marcadores moleculares tipo microsatélites o SSR. Se generaron 25 grupos de ligamiento identificando 3 QTL mayores asociados con una región del genoma de yuca con el contenido de carotenos totales. Tres marcadores SSR explicaron el 37.2% (NS109), 32% (rSSRY251) y 27.7% (rSSRY313) de la varianza fenotipica total, situados en el grupo de ligamiento D fuertemente asociados con el contenido de carotenos totales en la familia AM320 S1, se estableció una fuerte correlación positiva entre color de pulpa de raíz y contenido de carotenos totales (r=0.81), y negativa entre carotenos totales y materia seca (r= -0.31). Los tres posibles QTL presentaron efecto positivo y de carácter aditivo para el contenido de carotenos totales, pueden ser la oportunidad para implementar selección asistida por marcadores para carotenos totales en yuca.
Palabras claves: Manihot esculenta Crantz; caroteno; QTL; SSR.
ABSTRACT
The S1 population of the Thai variety MTAi8 (AM320), which shows patterns of segregation defined by the content of total carotenes (beta-carotene), underwent an analysis of segregating bulk (BSA = Bulk segregating Analysis) using 700 molecular markers type microsatellites or SSR. There were generated 25 tying groups and identifying 3 major QTLs associated with a region of the genome of cassava with the content of total carotenes. Three markers SSR; explained the 37.2% (NS109), 32% (rSSRY251) and 27.7% (rSSRY313) of the total phenotypic variance, placed in the Group D strongly tying, associated with the content of total carotenes in the family AM320 S1, which established a strong positive correlation between color and content root pulp carotenes total (r = 0.81), and between negative and carotenes total dry matter (r = -0.31). The three possible QTLs showed positive and additive effect for the content of totals carotenes and could be an opportunity to implement assisted selection markers for total carotenes in cassava.
Key words: Manihot esculenta Crantz; carotene; QTLs; SSR-microsatellites.
La yuca (Manihot esculenta Crantz), alimento básico en América Latina desde el período precolombino y que se introdujo en áfrica desde el siglo XVi (FAO, 2000), es una fuente pobre de macronutrientes y algunos micronutrientes, en particular de vitamina A (Kawano, 2003) pero aporta vitamina C, B2, B6, magnesio y potasio.
El contenido de betacaroteno es precursor clave en la síntesis de la vitamina A, además es característica de alta heredabilidad y se indica mediante el color de la raíz. El mapeo genético y de QTL son análisis básicos para entender el comportamiento genético de esta característica. De tal manera se pretende abordar el mejoramiento del contenido de caroteno y aprovechar la alta variabilidad genética existente en yuca a través de la vía convencional y mediante métodos de transformación genética (Fregene et al., 1997).
Los objetivos de la investigación fueron construir un mapa genético del contenido de carotenos totales; encontrar marcadores fuertemente asociados con el contenido de caroteno en yuca, y establecer regiones en el genoma (QTL) que tengan relación con el contenido de caroteno.
La investigación se adelantó en CiAT (Palmira, departamento del Valle del Cauca, a 3°16' norte, 76°32' oeste y 965 m.s.n.m.) con la población S1 producto de una autofecundación de la variedad tailandesa MTAi-8.
El experimento se sembró en 2005 y 2007, en un diseño de bloques completamente al azar con tres tratamientos y dos replicaciones, en parcelas simples de seis plantas por surco, distanciadas a 0.8 m y 1 m entre filas. Las variables materia seca, contenido de carotenos totales y color de raíz se evaluaron a los once meses.
El aislamiento de DNA se hizo con el protocolo de miniextracción (Dellaporta, 1983). Se realizó análisis de agrupamiento segregante (BSA) (Michelmore et al., 1991), lo cual generó dos agrupamientos para raíz naranja (10 genotipos) y blanca (10 genotipos). Los DNA de los agrupamientos y los padres se genotipificaron con 700 marcadores SSR viables para yuca (Mba et al., 2001). En aquellas evaluaciones en donde el polimorfismo se mantenía en los individuos, al igual que en los agrupamientos, se evaluaban los marcadores en toda la familia, población S1 AM320 (TAi8) conformada por 229 plantas. 140 SSR polimórficos se sometieron a análisis de mapeo genético y de QTL.
El análisis de ligamiento y mapeo genético se realizó usando un LOD score de 4.0 y una fracción de recombinación de 0.3 (Fregene et al., 1997). El mapa genético se construyó con 140 microsatélites, con el programa de computador Mapmaker 2.0 corrido sobre un Macintosh Centris 650 y una versión Mapmaker 3.0 (Lander et al., 1987). Para el mapeo de QTL en la población segregante S1 se usó análisis de marcador simple con el paquete software QGENE (Nelson, 1997). Un QTL se estableció cuando la significancia fue P < 0.005. Los resultados de R2 representan la varianza fenotípica total explicada. Para el análisis de QTL se usó el modelo estadístico de prueba de T (Liu y Liu, 1995) en el cual tres o más marcadores ligados se sometieron a análisis de regresiones múltiples.
Para el análisis de QTL se estableció un estudio constituido por tres modelos: A.S.M = Análisis de simple marcador; A.M.i = Análisis de mapeo por intervalo; A.M.i.C = Análisis de mapeo por intervalo compuesto. Este tipo de análisis establece tres categorías para la veracidad de un QTL en una región del genoma. Para el mapeo de QTL se usó mapeo de intervalo empleando un algoritmo de LOD > 3.0 con el paquete de computador Mapmaker/QTL y QGENE.
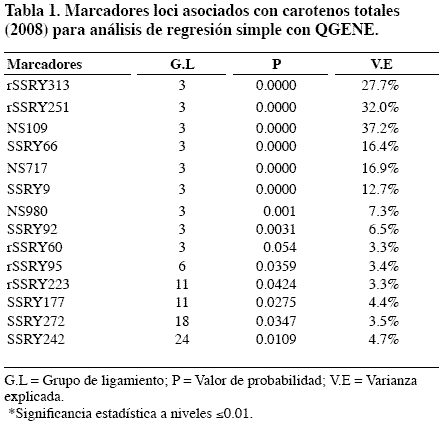
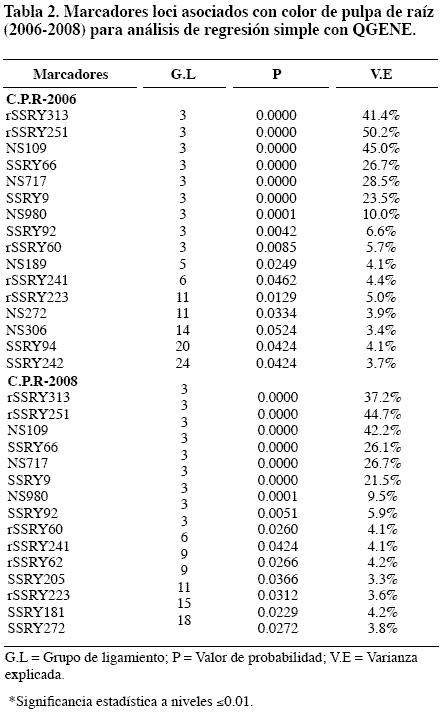
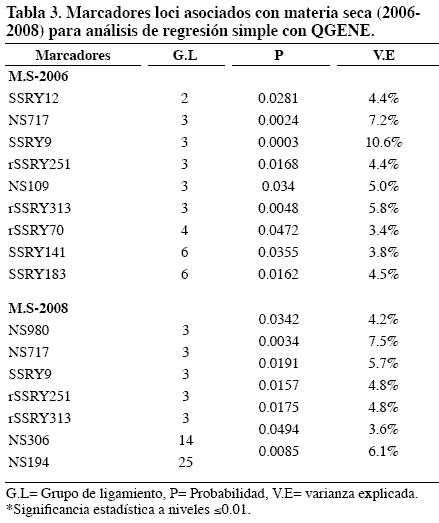
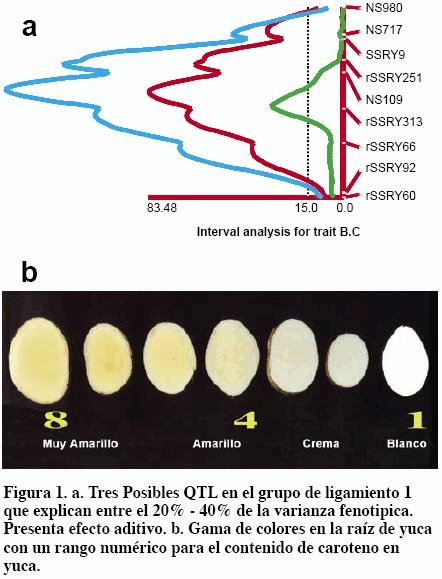
Se encontraron 17 posibles QTL en los grupos de ligamiento (1, 2, 3, 4, 6, 11, 15, 16, 19, 22) (Tabla 1),(Tabla 2),(Tabla 3) pero sólo tres QTL para CCT-2008 (contenido de carotenos totales); cuatro QTL para CPR-2006-2008 (color pulpa de raíz), situados en el grupo de ligamiento 1 presentaron significancia de P < 0.001. El grupo de ligamiento 1 tuvo LOD 33 para CCT-2008; LOD 68 para CPR-2006 y LOD 78 para CPR-2008 (Figura 1a), tres QTL explicaron 32.3% de la varianza fenotípica para contenido de carotenos totales, cuatro QTLs que afectaron el color de pulpa de raíz -2006 explicaron 41.3% y cuatro QTL para color de pulpa de raíz -2008 explicaron 37.7% (Tabla 4).
El mapa genético, que formó 25 grupos de ligamiento, cubrió 1476.6 cM con promedio entre marcadores de 21.5 cM (Figura 2), 37 marcadores SSR estuvieron desligados. 12 microsatélites (11.6%) segregaron normalmente con significancia de 0.001, 43 microsatélites (41.7%) con significancia de 0.01, 22 microsatélites (21.3%) con segregación de 0.05 de significancia y 26 microsatélites (25.2%) no segregaron según el valor esperado 1:2:1, sólo 12 grupos segregaron normalmente. Los casos más extremos de distorsión en la segregación en la población S1 se observaron sobre el marcador NS980 en el G.L 1, donde sólo 5 plantas de 229 fueron homocigotas para B.
Para construir el mapa también se utilizaron los 26 microsatélites que no segregaron según lo esperado, debido a que existen factores biológicos como marcador sistemático, error en la clasificación del genotipo, factores de rearreglos cromosómicos (Gebhardt et al., 1991; Kianian y Quiros, 1992), defectos de genes y efecto de dos o más genes recesivos que producen letalidad (Liu, 1998). Además, la segregación se debe a selección natural durante la fertilización, crecimiento de la planta, gametogénesis y desarrollo de semillas (Jung et al., 1996; Xu et al., 1997; Nacagahra, 1972; Harushima et al., 1996; Garris et al., 2003), factores que influyen en que las proporciones esperadas según las leyes de Mendel no sean como los valores establecidos para la segregación en una población S1 1:2:1. En la composición del genoma de la población S1 se presentaron 27% de individuos homocigotos para el alelo (A), 46.3% heterocigotos (H) y el 26.6% correspondió a individuos homocigotos con alelo (B).
Tres QTL se detectaron sobre el grupo de ligamiento 1; en donde se encuentran 6 SSR los cuales explican: 27.7% (rSSRY313), 32% (rSSRY251), 37.2% (NS109), 16.4% (SSRY66), 16.9% (NS717), 12.7% (SSRY9) de la varianza fenotípica total. Uno de los marcadores fuertemente asociado con el contenido de carotenos totales fue el SSRY251, el cual explica el 32% de la varianza fenotípica, la segregación de este marcador y la evaluación del color (Figura 1b, Figura 3a y Figura 3b), presenta alta correlación con el contenido de caroteno (r > 0.8). Se puede observar que según la intensidad del color, cuando el rango del color es 8, está asociado con la homocigocidad del tamaño pequeño del alelo para el SSRY251 en esta familia. Los tres posibles QTL se encuentran en el grupo de ligamiento 1 entre los intervalos: QCCT1 (rSSRY66-rSSRY313); QCCT2 (rSSRY313-NS109); QCCT3 (NS109-rSSRY251), los cuales pertenecen al grupo D del mapa genético de yuca (Fregene et al., 1997). Se podría decir que una región del genoma de yuca está asociada con el contenido de carotenos.
La varianza fenotípica total para la población mapeada S1 fue del 32.3% para la característica carotenos totales y 39.5% para color pulpa de raíz. Se presentaron 3 QTL mayores para CCT (contenido de carotenos totales): QCCT1, QCCT2, QCCT3 y 4 QTL para CPR (color pulpa de raíz): QCPR1, QCPR2, QCPR3, QCPR4 con efecto aditivo, siendo estos los componentes genéticos más importantes de un QTL para explicar la varianza fenotípica. Gracias a este tipo de población S1 se pudo determinar el efecto de diferentes estados del gen sobre el fenotipo, ya que estuvieron representados los tres posibles estados del gen.
Cinco probables QTL de segundo orden afectaron todas las características (CCT, CPR, MS), lo que indica que se observaron regiones de grupos ligados con múltiples QTL para características correlacionadas. Algunos de los QTL identificados en más de una característica coinciden en la región del genoma y en la dirección del efecto fenotípico, al sugerir pleiotropismo, tipo de efecto que puede ser responsable de las correlaciones negativas y positivas entre las tres características; la correlación negativa resulta de efecto pleiotrópico que afecta CCT-MS; CPR-MS (-0.31; -0.31 < 0.01) y correlación positiva CCT-CPR (0.81 < 0.01).
Las interacciones producidas entre QTL de primero y segundo orden (interacciones digénicas) se pueden explicar por factores epistáticos, ya que es posible que algunos de los QTL para las características que componen carotenos totales pueden tener efecto de comportamiento como factores modificantes y contribuir a la correlación genotípica. La epistasis produce erosión genética y reduce el vigor en especies relacionadas. En muchos estudios se observó que la producción de la progenie de cruces entre padres de la misma especie o subespecie es más baja que la de los padres (Li et al., 1996). Para este estudio los efectos de epistasis producidos entre muchas interacciones digénicas pueden ser una buena base genética para la característica de caroteno en el cultivo de yuca. Los resultados de interacciones digénicas tendieron a incrementar el contenido de caroteno.
La población S1 empleada en este estudio puede ayudar de forma más efectiva para discernir múltiples QTL sobre un mismo grupo de ligamiento, lo cual afecta la misma característica. Además, con este tipo de población se pueden distinguir factores aditivos de los dominantes. Como es probable que la población S1 sea engañosa por la considerable variación en características cuantitativas, especialmente cuando hay máximo desequilibrio de ligamiento, es necesario seguir estudiando estos efectos en generaciones recombinantes para determinar con mayor confiabilidad el efecto positivo, neutral o negativo producido por un alelo y determinar si se basa en interacciones digénicas sumado al análisis en diferentes ambientes.
Dos genes están involucrados en la biosíntesis de betacaroteno: phytoene sintasa y phytoene desaturasa (Hirschberg, 1998). Correa et al. (2004), utilizando la misma población S1 (AM320), con la variedad amarilla MTAi8, mapearon el phytoene syntasa el cual explicó 30% de la varianza fenotípica y se situó en un grupo de ligamiento diferente (5 para S1 y G para la F1 en yuca) al posible QTL asociado con los marcadores SSR de cDNA establecidos en este estudio. Por el contrario, el análisis de regresión simple entre phytoen syntasa, como variable independiente, y el contenido de betacaroteno, como variable dependiente, no mostró asociación con el gen que explicó 30% de la varianza fenotípica para el contenido de betacaroteno. Los resultados sugieren la existencia de otros genes que actúan sobre la herencia del contenido de betacaroteno en yuca.
QTL identificados y ligados a carotenos totales en yuca pueden ser de gran ayuda para incorporar en un esquema de selección asistida por marcadores en el mejoramiento de características de tipo micronutrientes relacionados con la salud humana. También MAS para carotenos totales pueden contribuir a identificar y seleccionar genotipos superiores, con el fin de reducir costos y tiempo en el programa de mejoramiento.
Se construyó el mapa genético para el contenido de carotenos totales en una población S1 de yuca usando marcadores "microsatélites", el cual sirvió de base para aplicar el análisis de QTL.
Siete marcadores SSR (rSSRY313, rSSRY251, NS109, SSRY66, NS717, SSRY9, NS980) situados en el grupo de ligamiento 1 y asociados con el contenido de carotenos totales en la familia S1 AM320 explicaron entre 30% y 40% de la varianza fenotípica.
Se logró detectar y localizar 3 QTL mayores para carotenos totales, con la respectiva acción y efecto sobre la característica.
Al proyecto "Harvest Plus" del CiAT que financió la tesis de maestría del biólogo J. A. Marín C., de la cual se derivó este artículo. Al doctor Martin Fregene por creer en este trabajo. A los integrantes del Laboratorio de Genética de yuca, muy especialmente a Janneth Gutiérrez, Adriana Alzate, Adriana Núñez y Diana Falla. A los trabajadores de campo del programa genética de yuca, Jairo Valencia, Armando Bedoya, Antonio López por su colaboración y asesoría.
Al laboratorio de Bioquímica del CiAT, especialmente a Alba Lucía Chávez, por la colaboración en los análisis de datos fenotípicos y carotenos totales.
1. Correa, A.; Barrera, E.; Castelblanco, W.; Morante, N.; Ceballos, H.; Tohme, J.; Fregene, M. 2004. Genetic Mapping of Genes Envolved in the Biosíntesis of Beta-carotene. inform Annual. Cali, Colombia: CiAT. p30-38. [ Links ]
2. Dellaporta, S.L.; Wood, J.; Hicks, J.R. 1983. A plant DNA minipreparation: version ii. Plant Mol Biol. Rep 1: 19-21. [ Links ]
3. FAO, 2000. La economía mundial de la Yuca, hechos tendencias y perspectivas. Roma-italia. p32-37. [ Links ]
4. Fregene, M.A.; Angel, F.; Gómez, R.; Rodríguez, F.; Roca, W.; Tohme, J.; Bonierbale, M. 1997. A molecular genetic map of cassava (Manihot esculenta Crantz). Theor Appl Genet. 95 (3): 431-441. [ Links ]
5. Garris, A. J.; Mccouch, S.R.; Kresovich, S. 2003. Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L). Genetics 165: 759-769.. [ Links ]
6. Gebhardt, C.; Ritter, E.; Barone, A.; Debener, T.; Walkemeier, B.; Schachtschabel, U.; et al. 1991. RFLP maps of potato and their alignment with the homoelogus tomato genome. Theor Appl Genet 83(1): 49-57. [ Links ]
7. Harushima, Y.; Kurata, N.; Yano, M.; Nagamura, Y.; Sasaki, T.; Minobe, Y.; et al. 1996. Detection of segregation distortions in an indica-japonica rice cross using a high-resolution molecular map. Theor Appl Genet 92 (2): 145-150. [ Links ]
8. Hirschberg, J. 1998. Molecular biology of carotenoid biosynthesis. p177-180. in: Britton, G.; Liaaen-Jensen, S.; Pfander, H. (eds). Carotenoids. Vol 3. Berlin: Birkhäuser Verlag. [ Links ]
9. Jung, G.; Coyne, P. W.; Shroch, J.; Nienhuis, E.; Arnaud-Santana, J.; Bokosi, H. M.; et al. 1996. Molecular markers associated with plant architecture and resistance to common blight, web blight, and rust in common beans. J Am Soc Hort Sci 121 (5): 794-803. [ Links ]
10. Kawano, K. 2003. Thirty years of cassava breeding for productivity – biological and social factors for success. Crop Sci 43 (4): 1325-1335. [ Links ]
11.Kianian, S. F.; Quiros, C. F. 1992. Generation of a Brassica Oleracea composite RFLP map: linkage arrangements among various populations and evolutionary implications. Theor App Genet 84(5-6): 544-554. [ Links ]
12. Lander, E.S.; Green, P.; Abrahamson, J.; Barlow, A.; Dal, M. J.; Lincoln, S. E.; Newberg, L. 1987. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174-181. [ Links ]
13. Li, Z. K.; Pinson, R. M.; Park, W. D.; Paterson, A. H.; Stansel, J. W. 1996. Epistasis for Three Grain Yield Components in Rice (Oryza sativa L). Genetics 145: 453-465. [ Links ]
14. Liu, B. H.; Liu, Y. Y. 1995. PGRi, a software for plant genome research. Plant Genome iii. Conference abstract, p 105. [ Links ]
15. Liu, B. 1998. Statical genomics. Linkage, Mapping and QTL analysis. Boca Raton: CRC Press. 562p. [ Links ]
16. Mba, R.; Stephenson, P.; Edwards, K.; Melzer, S.; Mkumbira, J.; Gullberg, U.; et al. 2001. Simple Sequence Repeat (SSR) markers survey of the cassava (Manihot esculenta Crantz) genome: towards an SSR-based molecular genetic map of cassava. Theor Appl Genet 102(1): 21-31. [ Links ]
17. Michelmore, R. W.; Paran, I.; Kesseli, R. V. 1991. identification of markers linked to disease – resistant genes by bulked segregant analysis: a rapid method to detect markers in specific genome regions by using segregating populations. Proc Natl Acad Sci USA 88: 9828-9832. [ Links ]
18. Nacagahra, M. 1972. Genetic mechanism on the distorted segregation of markers gene belonging to the eleventh linkage group in cultivated rice. J Breed. 22: 232-238. [ Links ]
19. Nelson, J. C. 1997. Q-gene: software for marker based genome analysis and breeding. Mol Breed 3: 229-235. [ Links ]
20. Xu, Y.; Zhu, L.; Xiao, J.; Huang, N.; Mccouch, S. R. 1997. Chromosomal regions associated with segregation distortion of molecular markers in F2, backcross, doubled haploid and recombinant inbreed populations in rice (Oyza sativa L). Mol Gener Genet 253: 535-545. [ Links ]

