










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Facultad de Medicina Veterinaria y de Zootecnia
Print version ISSN 0120-2952
Rev. Med. Vet. Zoot. vol.58 no.1 Bogotá Jan./Apr. 2011
HIPOADRENOCORTICISMO PRIMARIO CANINO: REPORTE DE CASO
CANINE PRIMARY HYPOADRENOCORTICISM: CASE REPORT
J. L. Granados,* L. M. Martínez, V. Galindo
Clínica para Pequeños Animales, Facultad de Medicina Veterinaria y de Zootecnia,
Universidad Nacional de Colombia, Bogotá (Colombia).
*Autor para correspondencia: jlgranadoss@unal.edu.co
Artículo recibido: 10 de octubre de 2009; aprobado: 16 de mayo de 2011
RESUMEN
Un canino macho Cocker Spaniel de 6 meses de edad fue presentado a la Clínica para Pequeños Animales de la Universidad Nacional de Colombia, con historia de vómito y diarrea de una semana de duración. El paciente presentaba hiperkalemia, y los electrocardiogramas realizados fueron compatibles con este hallazgo. El diagnóstico de hipoadrenocorticismo primario fue confirmado mediante la realización de una prueba de estimulación con ACTH. Posteriormente a la muerte súbita del paciente, el examen microscópico de las glándulas adrenales reveló cambios histológicos compatibles con la enfermedad de Addison. El hipoadrenocorticismo primario es un desorden endocrino poco común que afecta principalmente pacientes caninos; está caracterizado por la destrucción inmunomediada de las cortezas adrenales, lo cual conduce a deficiencia de glucocorticoides, mineralocorticoides y hormonas sexuales adrenales. La historia de los pacientes afectados es variable y los signos clínicos son usualmente inespecíficos; las pruebas de laboratorio, por lo general, revelan hiperkalemia e hiponatremia, resultantes de la pérdida de la secreción de aldosterona. La historia, el examen clínico, la ecografía abdominal y los hallazgos de laboratorio pueden indicar enfermedad de Addison, sin embargo, la prueba de estimulación con hormona adrenocorticotrópica (ACTH) es considerada como la prueba de oro para el diagnóstico definitivo de la entidad. La enfermedad de Addison no tiene cura, pero puede ser manejada con terapia médica para reemplazar las deficiencias de mineralocorticoides y glucocorticoides.
Palabras clave: hipoadrenocorticismo primario, enfermedad de Addison, glucocorticoides, mineralocorticoides.
ABSTRACTSix month old, male Cocker Spaniel was presented to the Small Animal Clinic at the Universidad Nacional de Colombia with a one week history of vomiting and diarrhea. The patient was hyperkalemic and the electrocardiogram results were consistent with this finding. The diagnosis of primary hypoadrenocorticism was confirmed by performing an ACTH stimulation test. Following the patient's sudden death, micros copic examination of the adrenal glands revealed histologic changes consistent with Addison`s disease. Primary hypoadrenocorticism is an uncommon endocrine disorder that primarily affects canine patients. The disorder is caracterized by the immune-mediated destruction of the adrenal cortices, resulting in mineralocorticoid, glucocorticoid and adrenal sex hormone deficiencies. Patient history is variable and clinical signs are often nonspecific; laboratory testing commonly reveals hyperkalemia and hyponatremia resulting from lack of aldosterone secretion. Clinical history, physical exam, abdominal ultrasound and laboratory findings may indicate Addison disease, however, the adrenocorticotrophic hormone (ACTH) stimulation test is considered to be the gold standard for definitive diagnosis of primary hypoadrenocortisism. Addison disease is not curable but can be managed with pharmaceutical therapy that replaces the mineralocorticoids and glucocorticoids deficiency.
Key words: primary hypoadrenocorticism, Addison disease, glucocorticoids, mineralocorticoids.
INTRODUCCIÓN
El hipoadrenocorticismo primario, o enfermedad de Addison, es una condición poco común en perros, y extremadamente rara en gatos. La mayoría de casos ocurren en hembras jóvenes (70%) y de edad intermedia, y ha sido reportada en una gran variedad de razas (Peterson et ál. 1989; Sadek y Schaer 1996; Shaker et ál. 1988). La incidencia actual en un gran número de hospitales animales ha sido calculada en 0,36 caninos por cada 1.000; se ha planteado que este pequeño número se debe al subdiagnóstico de la entidad (Grooters 1998).
Usualmente, la historia de los perros afectados es vaga e inespecífica con un curso fluctuante. Por lo general, la enfermedad es crónica y progresiva, sin embargo, está descrita la ocurrencia de crisis súbitas agudas, que cursan con hipovolemia, azotemia prerrenal, arritmias cardiacas, severa depresión mental, debilidad muscular, shock y muerte (Deborah y Greco 2007; Feldman y Nelson 2007; Kintzer y Peterson 1997; Reusch 2000; Willard et ál. 1982). Muchos pacientes tienen una historia de resolución de los signos clínicos solamente con terapia de fluidos intravenosos, pero tiempo después los signos clínicos recurren (Roth y Tyler 1999). La historia puede incluir la ausencia de respuesta a tratamientos previos para desórdenes gastrointestinales y renales; los signos a menudo sugieren desórdenes comunes, especialmente renales, gastrointestinales y enfermedades infecciosas. El tratamiento incluye el manejo de la crisis addisoniana y la suplementación de glucocorticoides y mineralocorticoides de por vida (Nelson 2005; Deborah y Greco 2007) como se describirá más adelante.
DESCRIPCIÓN DEL CASOA la Clínica para Pequeños Animales de la Facultad de Medicina Veterinaria y de Zootecnia, Universidad Nacional de Colombia, fue presentado un canino Cocker Spaniel macho, de 6 meses de edad, con historia de vómito recurrente, diarrea, anorexia y micción frecuente; previamente había sido atendido en otro centro veterinario con signos gastrointestinales y sin respuesta a los tratamientos instaurados.
En el examen clínico se observó depresión, bradicardia de 72 pulsaciones por minuto, tiempo de llenado capilar menor a 2 segundos, dolor a la palpación abdominal y evidencia de diarrea en el periné. Presentó dos episodios de vómito líquido, espumoso y amarillo durante la consulta. Se plantearon como diagnósticos diferenciales: gastroenteritis bacteriana (Campilobacter jejuni y/o Clostridium prefringens), viral (distemper canino y/o parvovirus), parasitaria (Toxocara canis, Ancylostoma sp. y/o Uncinaria sp.), cuerpo extraño intestinal, indiscreción alimentaria, pancreatitis y falla renal aguda.
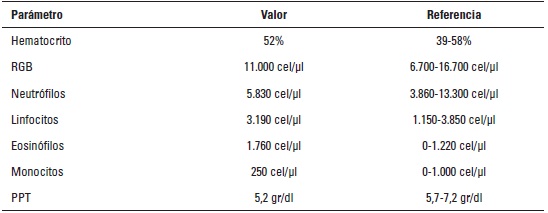
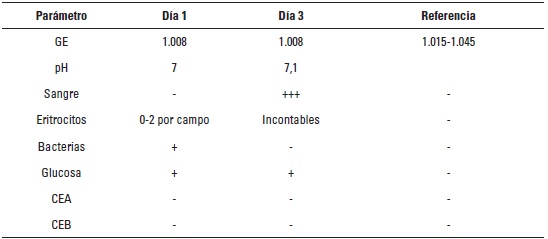
El hemograma (tabla 1) reportó de forma relevante eosinofilia (1.760 cel/μl) e hipoproteinemia (5,2 g/dl), además, presentó una medición de creatinina sérica elevada 309 μmol/L (tabla 2). En el urianálisis obtenido por sonda (tabla 3), se encontró una gravedad específica de 1.008, glucosa +, eritrocitos 0 a 2 por campo y bacterias escasas. Se midió la producción de orina en 24 horas, con un resultado normal (1,66 ml/k/hora [1-2 ml/k/hora]). En el coprológico presentó moco +++, sangre oculta ++++, cocos gram positivos ++ y bacilos gram positivos y negativos moderadamente aumentados.
TABLA 1. Hemograma del primer día, donde se observa eosinofilia e hipoproteinemia.
RGB: recuento de glóbulos blancos, PPT: proteínas plasmáticas totales
Se instauró fluidoterapia con solución salina fisiológica a razón de 120 ml/k/día, más la corrección de la deshidratación IV; dextrosa 50% 3 ml/k/día IV, ranitidina 2 mg/k SC cada 8 horas, metronidazol 15 mg/k IV cada 12 horas y ampicilina 20 mg/k IV cada 8 horas, Ensure® 340 kcal/día, según requerimientos energéticos basales ([30 (peso corporal en k)] + 70).
En los 5 días siguientes presentó vómito ocasional y el día 3 de evolución resolvió la diarrea; en un segundo urianálisis (tabla 3) realizado éste día se encontró GE de 1.008, glucosa +, presencia de sangre +++ y eritrocitos incontables. Se consideró diabetes mellitus debido a la glucosuria, y se realizó la glicemia 4,84 mmol/L (tabla 2), descartando así el diagnóstico diferencial de diabetes mellitus.
TABLA 2. Químicas sanguíneas obtenidas durante la hospitalización
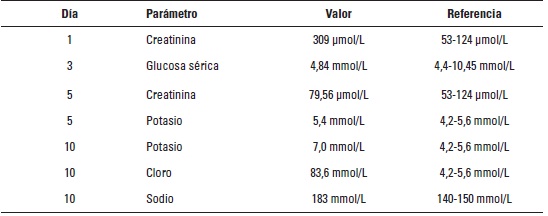
TABLA 3. Urianálisis de los días 1 y 3, donde se observa hipostenuria marcada, hematuria, piuria y glucosuria leves.
CEA: células epiteliales altas, CEB: células epiteliales bajas
El día 5 de evolución se encontró creatinina sérica normal 79,56 μmol/L (tabla 2), se realizó una ecografía abdominal sin observar hallazgos anormales, y un electrocardiograma (ECG) en el cual se observaron ondas T amplias y elevación del segmento S-T (> 0,2 mV) en la derivación II (figura 1); estos hallazgos son compatibles con hiperkalemia o hipoxia del miocardio, por lo que se determinó el potasio sérico: 5,4 mmol/L (tabla 2).
Por los signos clínicos y los hallazgos electrocardiográficos se incluyó un nuevo diagnóstico diferencial: enfermedad de Addison, aun a pesar de no haber encontrado una franca elevación en el nivel de potasio sérico. Se plantearon como planes diagnósticos un nuevo ECG, medición de sodio y potasio séricos, prueba de estimulación con ACTH, medición de cortisol en orina, relación creatininacortisol y una medición de cortisol sérico, tratamientos que por motivos económicos no se realizaron ese día.
Durante los días 6 a 9 de hospitalización el paciente estuvo alerta, sus constantes fisiológicas fueron normales, presentó vómito de manera ocasional y consumió alimento normalmente. Durante este periodo no se realizaron cambios en el manejo terapéutico del paciente.
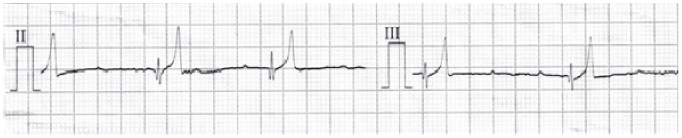
FIGURA 1. Electrocardiograma, derivadas II y III (25 mm/s, 10 mm/mV): se observa una disminución en la amplitud del complejo QRS,
aumento en la amplitud de las ondas T (superior al 25% de R) y elevación del segmento S-T de 0,3mV
El día 10 presentó dos episodios de vómito en la mañana, debilidad, anorexia, hipotermia (36,6 ºC), bradicardia (50 ppm), pulso débil y heces blandas. Se realizó medición de electrolitos séricos (tabla 2), donde se observó hiperkalemia, normocloremia e hipernatremia, con potasio sérico 7,0 mmol/L, cloro sérico 83,6 mmol/L, sodio sérico 183 mmol/L y relación sodio-potasio de 26:1 (> 27:1). Se mantuvieron los mismos planes terapéuticos y se adicionó metoclopramida 0,2 mg/k PO cada 8 horas, adicionalmente, ante la sospecha de hipoadrenocorticismo, se incluyó en la terapia fosfato sódico de dexametasona 0,5 mg/k IV cada 24 horas; en los días posteriores las constantes fisiológicas regresaron a la normalidad, el paciente presentó mejoría clínica, retomó el con-sumo de alimento, la debilidad cesó y presentó vómito esporádicamente.
El día 15 se realizó la prueba de estimulación con ACTH sintética Symantec® 0,25 mg intramuscular (IM), con cortisol basal previo: 0,22 μg/dl (0,1-6,15 μg/dl) y cortisol posestimulación: 0,02 μg/dl (6,0-15,2 μg/dl); así, el diagnóstico de hipoadrenocorticismo primario quedó confirmado y se planteó como terapia acetato de fludrocortisona (0,022 mg/k PO cada 24 horas) y prednisolona (0,22 mg/k PO cada 24 horas), sin embargo, restricciones económicas del propietario le impidieron adquirir la fludrocortisona. Durante la siguiente semana el animal se encontró alerta, con constantes normales y, aunque resolvió el vómito, consumió poco alimento.
El día 23, en horas de la mañana, el animal presentó heces blandas, bradicardia: 60 ppm y arcadas en forma ocasional. Se realizó un nuevo electrocardiograma (figura 2) y se encontró en la derivada II un aumento en el intervalo R-R y onda P ausente (pausa atrial), compatible con severa hiperkalemia; el paciente presentó colapso agudo y arresto cardiopulmonar.
El cadáver fue dispuesto para la necropsia, y como hallazgos macroscópicos relevantes, en las glándulas adrenales se observó disminución severa de tamaño, en la izquierda no había diferencia entre corteza y médula. Microscópicamente en las glándulas adrenales se reportó atrofia severa de las tres zonas corticales; reemplazo por tejido conectivo; cambios microcirculatorios; acumulación moderada de pigmentos y lípidos en macrófagos y libres; infiltrado difuso leve de mononucleares y células plasmáticas; en la médula adrenal, severa vacuolización celular. No se reportaron hallazgos anormales en el examen histopatológico de la hipófisis. De acuerdo con los hallazgos macroscópicos y microscópicos, se confirmó el diagnóstico de enfermedad de Addison.

FIGURA 2. Electrocardiograma realizado el día 23 (derivada II, 50 mm/s, 10 mm/mV): se observa microvoltaje,
amplitud similar de ondas R y T, complejos QRS prolongados y ausencia de ondas P (pausa atrial)
DISCUSIÓN
El paciente descrito en este reporte presentó los signos usuales de hipoadrenocorticismo como letargia, depresión, hipotermia, anorexia, vómito, debilidad, pérdida de peso, poliuria, polidipsia, dolor abdominal, deshidratación, temblores y bradicardia; otros signos clínicos comunes en la enfermedad, pero que no fueron presentados por este paciente, son melena y pulso femoral débil. Debido a la sintomatología inespecífica, el diagnóstico de hipoadrenocorticismo no fue considerado inicialmente, y se contemplaron otras enfermedades epidemiológicamente más frecuentes.
Debido a la bradicardia, se realizaron varios electrocardiogramas y se encontraron hallazgos característicos de hiperkalemia, muy usual en la enfermedad de Addison, junto con hiponatremia. En el paciente, la medición inicial de potasio sérico fue normal (5,4 mmol/L), y en mediciones posteriores se encontró notablemente por encima de ese valor (7,0 mmol/L); el valor inicial dentro de parámetros normales probablemente obedeció a un error analítico, pues en el momento de la toma de la muestra ya existían signos electrocardiográficos altamente sugestivos de hiperkalemia.
Existen anormalidades asociadas como leve a moderada hipocloremia, azotemia, hiperfosfatemia y acidosis metabólica (Feldman y Nelson 1996). Algunos pacientes atípicos tienen hiponatremia con normokalemia o hiperkalemia con normonatremia (Deborah y Greco 2007). El paciente presentó hiperkalemia, hipocloremia e hipernatremia; este último resultado fue dudoso, debido a que el valor de sodio estaba muy elevado y no era concordante con la hipocloremia presentada.
Algunos pacientes tienen mediciones electrolíticas normales, aún en presencia de hipocortisolemia. Sin embargo, la combinación de hiponatremia e hiperkalemia no está restricta a la insuficiencia adrenocortical; otros síndromes asociados con este patrón de electrolitos séricos en caninos incluyen varios desórdenes gastrointestinales, quilotórax y diabetes hiperosmolar no cetónica (DiBartola 1998; DiBartola et ál. 1985; Feldman y Nelson 1996; Roth y Tyler 1999; Willard et ál. 1991). Otras anormalidades descritas para la entidad incluyen hipercalcemia e hipoglicemia (Deborah y Greco 2007; Feldman y Nelson 2007; Kintzer y Peterson 1997; Reusch 2000; Roth y Tyler 1999).
La mayoría de pacientes presenta una relación sodio-potasio menor a 27:1; valores entre 27 y 20 deben ser considerados como sospechosos, mientras que por debajo de 20 son por lo general asociados con la enfermedad de Addison (Grooters 2002). En el paciente, esta relación fue de 26:1. Siempre debe descartarse hipoadrenocorticismo cuando se presentan hiperkalemia e hiponatremia, pero es posible que hasta 26% de los perros con la enfermedad tengan función mineralocorticoide normal y relaciones Na:K normales (Lifton et ál. 1996). Adicionalemente, los hallazgos clinicopatológicos de perros con enfermedades gastrointestinales primarias pueden ser similares a los del hipoadrenocorticismo, con relaciones Na:K por debajo de 27:1 y acidosis metabólica (Dibartola et ál. 1985).
La enfermedad está caracterizada por acidosis metabólica leve a moderada subsecuente al hipoaldosteronismo por la falla en la reabsorción renal de sodio en los túbulos distales, lo que reduce la habilidad para excretar H+ (Nelson 1980). Esta incapacidad para excretar protones, junto con la hipotensión e hipoperfusión tisular, explican la acidosis leve documentada en la mayoría de casos durante la crisis hipoadrenal. La medición de gases arteriales y dióxido de carbono venoso total, son métodos adecuados para determinar el estado ácido/ base en estos pacientes (Feldman y Nelson 2007).
El electrocardiograma es útil para la detección de las varias y complejas anormalidades asociadas con la hiperkalemia. Varias de las anormalidades electrocardiográficas descritas para la enfermedad fueron vistas en el paciente, incluyendo un incremento en la amplitud de ondas T, complejos QRS prolongados, intervalos P-R prolongados, ausencia de las ondas P y amplitud disminuida de las ondas R (Feldman y Nelson 1996; Kintzer y Peterson 1997; Tilley 1992). Se reporta que estas anormalidades electrocardiográficas no ocurren a menos que los niveles de potasio sérico excedan 7,5 mmol/L, y se ha visto que no ocurren en niveles inferiores a 7,0 mmol/L cuando el sodio sérico es menor a 130 mmol/L. Únicamente en casos severos de hiperkalemia, se justifica realizar terapia específica, la cual consiste en la administración de soluciones de gluconato de calcio al 10%, bicarbonato de sodio y/o soluciones dextrosa-insulina (Schaer 2001).
En el hipoadrenocorticismo primario se puede observar anemia normocítica normocrómica, asociada con una mínima respuesta de producción de reticulocitos; esto se debe a la combinación de efectos de mielosupresión asociados con la enfermedad crónica y la pérdida de sangre en el tracto gastrointestinal. Otros hallazgos incluyen linfocitosis y eosinofilia, atribuidas a la hipocortisolemia; sin embargo, su ocurrencia en perros con hipoadrenocorticismo es rara, y no es un indicador sensible del desorden (Feldman y Nelson 1996; Oelkers 1996). En este paciente se encontró eosinofilia absoluta persistente en todos los hemogramas; la ausencia de leucograma de estrés en un animal estresado como el aquí descrito, es un hallazgo común (Feldman y Nelson 1996; Kintzer y Peterson 1997; Reusch 2000; Roth y Tyler 1999) y puede orientar hacia el diagnóstico.
Es común observar azotemia prerrenal en pacientes con hipoadrenocorticismo primario (Peterson et ál. 1996); este paciente presentó una medición inicial elevada de creatinina sérica que regresó a parámetros normales después del inicio de la fluidoterapia intravenosa, lo que confirma la azotemia prerrenal.
El urianálisis de estos pacientes es usualmente hipostenúrico, como se observó en este individuo, lo que es atribuido a las pérdidas renales de sodio y el subsecuente lavado medular renal, aunque también podría sugerir falla renal (Tyler et ál. 1987).
La ultrasonografía abdominal puede revelar una reducción en el tamaño de las glándulas adrenales (Hoerauf y Reusch 1999), sin embargo, no fue posible localizar las glándulas adrenales del paciente en las ecografías realizadas.
El diagnóstico definitivo del hipoadrenocorticismo depende de la demostración de secreción mínima o ausente de cortisol desde la corteza adrenal en respuesta a la inyección de corticotropina (ACTH) (Feldman y Nelson 1996; Peterson et ál. 1996; Schaer 2001). En el paciente se realizó la prueba de estimulación con ACTH, en la cual se obtuvieron mediciones subnormales de cortisol antes y después de la aplicación de ACTH 0,22 μg/dl y 0,02 μg/dl, respectivamente, lo cual confirma el diagnóstico de enfermedad de Addison. Niveles sanguíneos < 1,0 μg/dl caracterizan la gran mayoría de perros con insuficiencia adrenal (Feldman y Nelson 2007). Sin embargo, un paciente se considera como positivo si se obtiene cortisol < 2 μg/dl; valores entre 2,5 y 5 μg/dl son dudosos (Schaer 2001), aunque se reporta que un perro normal tendrá un nivel de cortisol basal entre 0,1-6,15 μg/dl, y posestimulación de 6,0-15,2 μg/dl después de 60 minutos de aplicada la inyección de ACTH (Feldman y Nelson 1996). Deben descartarse otras causas de supresión adrenocortical que interfieran con la prueba de estimulación con ACTH, como por ejemplo, los tratamientos prolongados con glucocorticoides. El nivel de ACTH plasmático puede ser obtenido para distinguir entre hipoadrenocorticismo primario y secundario; la ACTH se encuentra alta en la enfermedad primaria, y más baja de lo normal en la secundaria. Esta prueba no se considera usualmente necesaria para iniciar un tratamiento apropiado. Además, la ACTH es extremadamente lábil, y requiere un manejo especial de las muestras, que hará que los resultados sean a menudo no fiables (Feldman y Nelson 1996; Kintzer y Peterson 1999; Peterson et ál. 1996).
El manejo a largo plazo de la enfermedad debe realizarse reemplazando los glucocorticoides y mineralocorticoides deficientes. El mineralocorticoide más utilizado es el acetato de fludrocortisona, 0,01-0,02 mg/k PO cada 12 horas; generalmente se requiere emplear la dosis más alta durante los primeros 6 meses de tratamiento. También puede ser usado el pivalato de desoxicorticosterona (DOCP), 2 mg/k IM o SC una vez cada 25 días aproximadamente, dependiendo del caso (Feldman y Nelson 1996; Hoerauf y Reusch 1999; Kintzer y Peterson 1999; Peterson et ál. 1996).
Como glucocorticoide puede emplearse prednisolona 0,1-0,2 mg/k PO cada 24 horas, inicialmente. La mayoría de los perros tratados con fludrocortisona no necesitan prednisolona debido a la actividad glucocorticoide del producto. En contraste, los perros tratados con DOCP requieren adicionalmente el tratamiento con glucocorticoide. La dosis de glucocorticoide administrada debe disminuir hasta lograr la dosis mínima efectiva, e incrementar durante los periodos en los cuales el animal sea sometido a situaciones estresantes, como enfermedades, cirugías o viajes. Durante el inicio del tratamiento se deben monitorear los electrolitos cada semana hasta que el paciente se encuentre estable, y luego de esto, cada 3 a 4 meses (Feldman y Nelson 1996; Horeauf y Reusch 1999; Kintzer y Peterson 1999; Peterson et ál. 1996).
En este paciente no fue posible instaurar la terapia adecuada, debido a la imposibilidad del propietario para adquirir el fármaco requerido (fludrocortisona). Seguramente esto permitió el desarrollo de una crisis hipoadrenocortical aguda que lo llevó a la muerte.
En la literatura está bien descrita la presentación de crisis súbitas agudas, que cursan con hipovolemia, azotemia prerrenal, arritmias cardiacas, severa depresión mental, debilidad muscular, shock y muerte (Deborah y Greco 2007; Feldman y Nelson 1996; Kintzer y Peterson 1997; Nelson 2005; Reusch 2000; Willard et ál. 1982). Es muy probable que esta fuera la causa del curso cíclico de recuperaciones y recaídas del paciente, sin una causa evidente, y de su fallecimiento inesperado, ya que, a pesar de encontrarse aparentemente estable, no había recibido el tratamiento recomendado.
La descompensación o crisis addisoniana ocurre de forma aguda y sin signos previos de enfermedad; es una emergencia que requiere tratamiento inmediato, y debe realizarse la reposición de fluidos vía endovenosa, con cloruro de sodio al 0,9%, iniciando con una dosis de 20-40 ml/k/día, e ir aumentando gradualmente hasta 90 ml/k/día, de acuerdo con el monitoreo de los electrolitos. Este monitoreo se inicia 15 a 20 minutos después de instaurada la terapia, para evitar las posibles complicaciones ocasionadas por la rápida corrección de los niveles de sodio sérico, principalmente la mielinosis pontina central, la cual ha sido reportada en varias especies incluyendo el perro (Roth y Tyler 1999; Sadek y Schaer 1996; Schaer et ál. 1986). También se ha reportado edema pulmonar durante la fase inicial de administración rápida de fluidos IV, por lo cual deben monitorearse constantemente los pacientes en busca de signos de dificultad respiratoria, así como la medición de la presión venosa central. Después de las primeras 24 horas, puede mantenerse la hidratación a 60 ml/k/día en caninos adultos, y 120 ml/k/día en cachorros, hasta que el animal reanude el consumo de alimento y agua con una producción normal de orina, y cuando los valores de electrolitos y BUN se encuentren dentro de los parámetros normales. Usualmente esto ocurre entre las 48 a 72 horas siguientes al tratamiento (Schaer 2001). En casos de crisis addisoniana, la deficiencia de glucocorticoides debe ser corregida mediante la administración de medicamentos de acción rápida como el succinato sódico de metilprednisolona 5-10 mg/k, el fosfato de dexametasona 2-5 mg/k IV. Inicialmente debe ser evitada la administración oral en pacientes que presenten vómito, y en estos casos el uso de metoclopramida (0,2-0,4 mg/kg SC cada 8 horas) u ondansetrón (0,1-1 mg/k IV cada 12 horas) puede ser de utilidad. Los requerimientos subsecuentes de glucocorticoides pueden ser suplidos con la administración de prednisolona a 1 mg/k PO, IM o IV cada 12 horas durante el segundo día, y reducir la dosis a 0,25 o 0,5 mg/k durante los primeros días de hospitalización (Roth y Tyler 1999). La dexametasona no interfiere con los niveles de cortisol endógenos medidos en la prueba de estimulación con ACTH, por tanto, el tratamiento puede ser instituido con este medicamento antes de realizar la prueba (Feldman y Nelson 1996; Kintzer y Peterson 1997).
La acidosis metabólica está usualmente en el rango leve a moderada, y rara vez requiere tratamiento específico, sin embargo, durante la crisis addisoniana puede ocurrir acidosis severa (pH < 7,1; bicarbonato < 10 mmol/L). En estos casos se administra bicarbonato de sodio durante 2 a 6 horas según la fórmula 0,2 x peso corporal (k) x (concentración deseada de bicarbonato en el plasma —la concentración de bicarbonato plasmático del paciente—). Es necesario medir los gases sanguíneos de forma seriada después de la administración de bicarbonato, el objetivo de la terapia es aumentar el pH sanguíneo a 7,2 o la concentración de bicarbonato a 12 mmol/L (Grooters 2002). Si el estado ácido/base no puede ser valorado en un paciente con crisis hipoadrenocortical, no se recomienda la terapia con bicarbonato. El manejo adecuado con líquidos y mineralocorticoides debería restaurar la perfusión renal, la cual, a su vez, aumenta la excreción de protones en la orina. Típicamente esto evita la administración de bicarbonato parenteral (Feldman y Nelson 2007).
Una vez la crisis addisoniana es controlada, el pronóstico es excelente, con un promedio de vida de 7 años a partir del diagnóstico y comienzo del tratamiento; con la apropiada terapia de reemplazo glucocorticoide y/o mineralocorticoide, se debe esperar a que los animales tengan una vida normal.
Al propietario de un paciente con hipoadrenocorticismo se le debe hacer énfasis en la importancia de la terapia de por vida, y en la posible necesidad de aumentar las dosis de glucocorticoides durante situaciones estresantes, tales como hospitalización, viajes y cirugías (Feldman y Nelson 1996; Meeking 2007). Infortunadamente en el paciente aquí descrito no se inició la terapia específica para la enfermedad, lo cual muy probablemente permitió la ocurrencia de una crisis hipoadrenocortical que lo llevó a la muerte.
AGRADECIMIENTOSAl doctor Germán Pérez, quien colaboró activamente con el diagnóstico de este caso, y a todo el equipo de profesionales y estudiantes de la Clínica para Pequeños Animales, en especial a la doctora Pilar Patiño y al doctor Juan Lucas Vargas por su dedicación y por los cuidados brindados a Tommy.
REFERENCIAS
1. Deborah S, Greco D. 2007. Hypoadrenocorticism in small animals. Clin Tech Small Anim Pract. 22(1): 32-35.
[ Links ]2. DiBartola SP, Johnson SE, Davenport DJ, Prueter JC, Chew DJ, Sherding RG. 1985. Clinicopathologic findings resembling hypoadrenocorticism in dogs with primary gastrointestinal diseases. J Am Vet Med Assoc. 187(1): 60-63.
[ Links ]3. DiBartola SP. 1998. Hyponatremia. Vet Clin North Am Small Anim Pract. 28: 515-550.
[ Links ]4. Feldman EC, Nelson RW. 1996. Canine and feline endocrinology and reproduction. 2a ed. Philadelphia: W.B. Saunders Company. p. 437-485.
[ Links ]5. Feldman EC, Nelson RW. 2007. Endocrinología y reproducción canina y felina. 3a ed. Philadelphia: W.B. Saunders Company. p. 437-486.
[ Links ]6. Grooters AM. 1998. Addison's disease: diagnosis and treatment. En: Florida Eastern States Veterinary Association, editors. Proceedings of the North American Veterinary Conference; 1998 enero 10-14, Orlando, Florida. p. 238-242.
[ Links ]7. Grooters AM. 2002. Tratamiento con líquidos en los trastornos endocrinos y metabólicos. En: DiBartola, editor. Terapia de líquidos en pequeñas especies. 2a ed. Columbus, Ohio: McGraw-Hill Interamericana. p. 400-412.
[ Links ]8. Hoerauf A, Reusch C. 1999. Ultrasonographic evaluation of the adrenal glands in six dogs with hypoadrenocorticism. J Am Anim Hosp Assoc. 35(3): 214-218.
[ Links ]9. Kintzer PP, Peterson ME. 1997. Primary and secondary canine hypoadrenocorticism. Vet Clin North Am Small Anim Pract. 27(2): 349-357.
[ Links ]10. Lifton SJ, King LG, Zerbe CA. 1996. Glucocorticoid deficient hypoadrenocorticism in dogs: 18 cases (1986-1985). J Am Vet Med Assoc. 209(12): 2076-2081.
[ Links ]11. Meeking S. 2007. Treatment of acute adrenal insufficiency. Clin Tech Small Anim Pract. 22(1): 36-39.
[ Links ]12. Nelson DH. 1980. The adrenal cortex: phisiological function and disease. Philadelphia: WB Saunders.
[ Links ]13. Nelson RW. 2005. Enfermedades de la glándula adrenal. En: Couto CG, Nelson RW, editores. Medicina interna de animales pequeños. 3 ed. Buenos Aires: Inter-Médica. p. 820-858.
[ Links ]14. Oelkers W. 1996. Adrenal insufficiency. New Eng J Med. 335: 1206-1212.
[ Links ]15. Peterson ME, Greco DS, Orth DR. 1989. Hypoadrenocorticism in ten cats. J Vet Int Med. 3: 55-58.
[ Links ]16. Peterson ME, Kintzer PP, Kass PH. 1996. Pretreatment clinical and laboratory findings in dogs with hypoadrenocorticism: 225 cases (1979-1993). J Am Vet Med Assoc. 208(1): 85-91.
[ Links ]16. Reusch CE. 2000. Hypoadrenocorticism. In: Ettinger SJ, Feldman EC editores. Textbook of veterinary internal medicine. 5a ed. Philadelphia: W.B. Saunders Company. p. 1488-1498.
[ Links ]17. Roth L, Tyler RD. 1999. Evaluation of low sodium: potassium ratios in dogs. J Vet Diagn Invest. 11:60-64.
[ Links ]18. Sadek D, Schaer M. 1996. Atypical Addison's disease in the dog: a retrospective survey of 14 cases. J Am Anim Hosp Assoc. 32(2): 159-163.
[ Links ]19. Schaer M, Riley WJ, Buergelt CD, Bowen DJ, Senior DF, Burrows CF et ál. 1986. Autoimmunity and Addison's disease in the dog. J Am Anim Hosp Assoc. 22: 789-794.
[ Links ]20. Schaer M. 2001. The treatment of acute adrenocortical insufficiency in the dog. J Vet Emerg Crit Care. 11: 7-14.
[ Links ]21. Shaker E, Hurvitz AI, Peterson ME. 1988. Hypoadrenocorticism in a family of standard poodles. J Am Vet Med Assoc. 192(8): 1091-1092.
[ Links ]22. Tilley LP, editores. 1992. Essentials of canine and feline electrocardiography, interpretation and treatment. 3 ed. Philadelphia: Lea and Febiger.
[ Links ]23. Tyler RD, Qualls CWJ, Heald RD, Cowell RL, Clinkenbeard KD. 1987. Renal concentrating ability in dehydrated hyponatremic dogs. J Am Vet Med Assoc. 191(9): 1095-1100.
[ Links ]24. Willard MD, Schall WD, McCaw DE, Nachreiner RF. 1982. Canine hypoadrenocorticism: report of 37 cases and review of 39 previously reported cases. J Am Vet Med Assoc. 180(1): 59-62.
[ Links ]25. Willard MJ, Fossum TW, Torrance A. 1991. Hyponatremia and hyperkalemia associated with idiopathic or experimentally induced chylothorax in four dogs. J Am Vet Med Assoc. 199:353-358.
[ Links ]
