










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombian Journal of Anestesiology
Print version ISSN 0120-3347
Rev. colomb. anestesiol. vol.35 no.2 Bogotá Jan./Apr. 2007
ARTÍCULO DE REVISIÓN
Vasoespasmo cerebral secundario a hemorragia
subaracnoidea por ruptura de aneurisma intracerebral
Juan A. Mejía C.1, María C. Niño de Mejía2, Leopoldo E. Ferrer Z.3, Darwin Cohen M.4
1 Neurocirujano. Hospital Universitario. Fundación Santa Fe de Bogotá. HUSI
2 Neuroanestesióloga. Hospital Universitario. Fundación Santa Fe de Bogotá.
3 Anestesiólogo Intensivista. Hospital Universitario. Fundación Santa Fe de Bogotá
4Neuroanestesiólogo. Hospital Universitario. Fundación Santa Fe de Bogotá
RESUMEN
La enfermedad cerebrovascular (ECV) es la tercera causa de muerte en países industrializados. De todos los tipos de EVC, la hemorragia subaracnoidea (HSA) es responsable de 22% a 29% de la mortalidad. Alrededor de 30% de los pacientes con hemorragia subaracnoidea (HSA) secundaria a ruptura de aneurisma cerebral, desarrollan vasoespasmo arterial y con ello el déficit neurológico asociado aumenta. Esta complicación empeora el pronóstico de los pacientes, puesto que un 25% de ellos mueren y otro 30% a 35% sufren de déficit neurológico permanente.
Entonces, el vasoespasmo cerebral es el factor modificable más importante para mejorar la tasa de morbimortalidad en pacientes con hemorragia subaracnoidea secundaria a ruptura de aneurisma. Estos puntos nos obligan a definir esquemas de intervención bien desarrollados y esquematizados, en donde el primer objetivo sea la prevención, permitiendo hacer un diagnóstico ultra temprano y que incluya un esquema de intervención bien definido que pueda ayudar a frenar el curso de la devastadora historia natural de esta complicación.
Palabras clave: hemorragia subaracnoidea, vasoespasmo cerebral, ruptura de aneurisma intracerebral.
SUMMARY
The cerebrovascular disease (CVD) is the third cause of death in industrializad countries. Of all the types of CVD, the subarachnoid hemorrhage (HAS) is responsible from 22% to 29% of mortality. Around 30% of the patients with secondary subarachnoid hemorrhage (HAS) to rupture of cerebral aneurism develops arterial vasospasm and with it the associated neurological deficit increases. This complication the prediction of the patients, since 25% of them die and another 30 to 35% undergo permanent neurological deficit.
Then, cerebral vasospasm is the modified factor more important to improve the rate of morbidity and mortality in patients with secondary HAS dur to aneurism rupture. These points compel us to define schemes of developed intervention directed to prevention allowing to make do an early extreme diagnosis that includes defined schemes of intervention that help to restrain the course of the devastating natural history of this complication.
Key works: subarachnoid hemorrage arterial vasospasm, rupture of cerebral aneurism.
1. INTRODUCCIÓN
Los reportes del sistema de salud norteamericanos ubican a la enfermedad cerebrovascular (ECV) como la tercera causa de morbilidad y mortalidad en ese país, resultados que extrapolamos a nuestro medio, ya que no tenemos estadísticas confiables a este respecto1.
La hemorragia subaracnoidea (HSA) corresponde a 6-8% del total de ECV. Tiene una incidencia de 30.000 pacientes por año, y prevalencia mundial de 6 a16 por 100.000 habitantes; la más alta en países como Finlandia y Japón y ocasiona el 22- 29% de las muertes de causa cerebrovascular2-4.
La mortalidad en el primer año de pacientes con vasoespasmo se sitúa entre el 25 y el 50%, un déficit neurológico permanente se observa en otro 15 a 20% y sólo 30-35% tiene una recuperación moderada a buena en un año2 (tabla 1).

El vasoespasmo cerebral posterior a hemorragia subaracnoidea por ruptura aneurismática es la principal causa de muerte y de déficit neurológico permanente en supervivientes de HSA1,2,5 y, por tanto, es el factor más frecuentemente modificable para mejorar el resultado en este grupo de pacientes.
Se describe que 20 a 30% de los pacientes con HSA presentan vasoespasmo sintomático y que aún después de recibir terapia máxima, el 50% desarrollará infarto cerebral, otro 15 a 20% tendrán déficit neurológico permanente o morirán en el primer año de evolución2, 7.
2. DEFINICIÓN Y CLASIFICACIÓN
El vasoespasmo cerebral es una condición reversible que cursa con reducción del calibre de la luz de una arteria en el espacio subaracnoideo cerebral, con la consiguiente disminución del flujo sanguíneo a las áreas perfundidas por el vaso comprometido8. (figura 1).
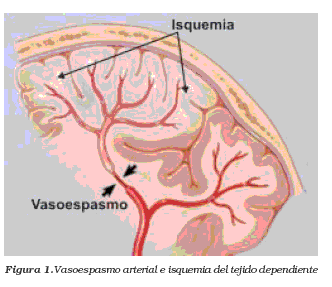
Se observa fundamentalmente después de HSA aneurismática, pero también se ha relacionado con trauma craneoencefálico y cirugía del área hipotalámico- hipofisiaria.
Se aceptan actualmente dos clasificaciones para vasoespasmo: angiográfico y sintomático.
2.1. Angiográfico: desde 1951 Ecker y Riemenschneider describen el vasoespasmo angiográfico como adelgazamiento de la columna de medio de contraste en las arterias cerebrales mayores1.
La clasificación angiográfica determina el porcentaje de reducción del diámetro del vaso, comparado con el observado en la angiografía inicial.
Se denomina vasoespasmo grave a un estrechamiento mayor de 75%; moderado de 50% a 75% y leve de 25% a 50%8,10.
El vasoespasmo angiográfico usualmente inicia del día 3° al 5o de la HSA, se observa la mayor estrechez de la luz arterial desde el día 5 al día 7, y se prolonga en promedio hasta el día 14; se resuelve en el curso de 2 a 4 semanas. Es observado en 30 - 70% de las arteriografías realizadas en el día 7 después de la HSA3. Es de notar que, al menos, buena parte del estrechamiento vascular sucede en vasos de pequeño calibre, que no son visibles con angiografía cerebral convencional y que pueden causar parte del compromiso neurológico del paciente10.
2. Sintomático: denominado también déficit neurológico tardío, se refiere al síndrome originado por el compromiso isquémico de una región cerebral, por estrechamiento de uno o varios vasos cerebrales, que origina la aparición de un nuevo deterioro del estado de conciencia, afasia o déficit motor1,211,12. (figura 1). Ha sido asociado con pronóstico neurológico muy pobre en supervivientes de HSA.
Dada la alta frecuencia de esta patología en HSA y la baja tasa de respuesta clínica a las terapias conocidas en la actualidad, el énfasis del tratamiento está no sólo en el diagnóstico temprano sino en la instauración de una terapia profiláctica como parte del manejo, que intente reducir el número de pacientes que desarrollen esta complicación.
3. FISIOPATOLOGÍA DEL VASOESPASMO
Este aspecto de la patología no esta claramente entendida. El estrechamiento de la luz del vaso puede causar aumento de la resistencia vascular y con ello disminución del flujo sanguíneo cerebral (FSC) a niveles críticos que pueden causar isquemia y lesión cerebral.
Es ampliamente aceptado que los componentes sanguíneos extravasados por la HSA contribuyen al vasoespasmo, dado que existe una relación clara entre el tiempo de ruptura eritrocitaria y la desaparición del coágulo en el espacio subaracnoideo, con el inicio y la finalización del vasoespasmo.
Los componentes sanguíneos liberados en el espacio subaracnoideo liberan productos metabólicos primarios o secundarios responsables del vasoespasmo clínico.
Se ha observado que existe un desequilibrio entre la real reducción del FSC y la tasa de utilización de glucosa; un estudio experimental encontró una exagerada tendencia a la anaerobiosis, con reducción en la taza de extracción de oxígeno, lo que puede contribuir al desarrollo de isquemia encefálica13.
Citaremos algunas de las teorías más aceptadas para explicar el origen del vasoespasmo después de HSA aneurismática.
3.1. Contracción arterial prolongada
Según esta teoría, el vasoespasmo resulta de una contracción prolongada del músculo liso, vascular, arterial, causado por la presencia de oxihemoglobina en el espacio subaracnoideo; ésta tendría efectos directos sobre la vasculatura y facilitaría la formación de radicales libres vasoconstrictores a partir de ella.
Los radicales superóxido se producen a partir del oxígeno cedido por la oxihemoglobina en el espacio subaracnoideo; al entrar en contacto con óxido nítrico del medio origina el radical peroxinitrito que ejerce efectos lesivos locales y decrementa los niveles perivasculares de óxido nítrico, con pérdida de la acción vasodilatadora de esta molécula.
La inactivación del óxido nítrico originó un aumento de la actividad de proteincinasa C que, a través de la movilización de depósitos de calcio intracelular mediado por calmodulina, es capaz de activar al complejo actina/miosina en la capa muscular lisa y con ello causar vasoconstricción. Este efecto se ve reforzado por un desequilibrio entre prostaglandinas vasoconstrictoras y vasodilatadoras en el medio local1.
3.2. Neuropéptidos vasoactivos
Se sugiere que la constricción arterial puede ser inducida por mecanismos de denervación e hipersensibilidad. En pacientes con hemorragia subaracnoidea se ha descrito disminución de catecolaminas y de otras sustancias, como neuropéptidos vasoactivos, en las terminales nerviosas perivasculares, con hipersensibilidad a las mismas14,15.
Las teorías que involucran a factores neurohormonales como causales de vasoespasmo arterial, se apoyan en la existencia de inervación para el árbol arterial cerebral de sistemas neuronales del nervio trigémino, que reciben señales producidas por la sangre liberada en el espacio subaracnoideo; estas señales aferentes son llevadas a núcleos catecolaminérgicos en el tallo, luego, a través de conexiones ascendentes se llevan señales al hipotálamo (principalmente, a la eminencia media) y, finalmente, esta estructura media en las respuestas que alteran el equilibrio neurohormonal y afectan el tono vascular activando los cambios vasoconstrictores observados en el vasoespasmo por HSA1,16.
Estudios experimentales en ratas encontraron que lesiones específicas en segmentos esenciales de la vía neural descrita, evitaban en todos los casos la aparición de vasoespasmo después de HSA16.
Algunos de los neuropéptidos estudiados como mediadores del vasoespasmo son el péptido relacionado con el gen de la calcitonina (PRGC), el neuropéptido Y y la sustancia P. Todas estas neurohormonas se encuentran almacenadas en las terminales nerviosas simpáticas perivasculares y pueden jugar un papel importante en la homeostasis del FSC, así como en el origen fisiopatológico del vasoespasmo cerebral17.
Un estudio experimental evaluó la posible participación del neuropéptido Y en este aspecto. Se de mostró en un modelo de estudio in vitro que es un poderoso vasoconstrictor arterial, que este efecto se prolonga en el tiempo, no depende de la función endotelial y es inhibido completamente con la administración de nifedipina, por lo que podría estar implicado en la patogénesis de la vasoconstricción retardada característica del vasoespasmo. Este mismo estudio también evaluó el papel del PRGC aplicado a arterias cerebrales in vitro; se observó que ejerce un poderoso efecto vasodilatador, de duración prolongada, mediado directamente por dilatación del músculo vascular liso17. Estos efectos no se ven alterados por la presencia de oxihemoglobina, que sí interfiere con el efecto vasodilatador del óxido nitroso y la prostaciclina1, lo cual demostró que su aplicación local funcionó al impedir la vasoconstricción retardada después de HSA17.
3.3. Cambios estructurales en la pared arterial
La contracción prolongada de la musculatura lisa de las arterias origina cambios morfológicos secundarios que típicamente consisten en hiperplasia de la íntima o fibrosis subendotelial, con arrugas en la membrana elástica interna y proliferación del tejido conectivo arterial18,19. En la luz, los glóbulos blancos y plaquetas se agregan y colaboran con el engrosamiento de la pared vascular. Los cambios estructurales resultantes de la hiperplasia arterial, agregación plaquetaria y edema llevan a incrementar la resistencia cerebrovascular y disminuyen el flujo sanguíneo cerebral1.
3.4. Respuesta inflamatoria
Esta teoría postula mecanismos inflamatorios, ya sean neurogénicos o asociados a la clásica cascada de inflamación, como iniciadores del proceso patológico.
3.4.1. Inflamación neurogénica
La producción antidrómica de sustancia P y del péptido relacionado con el gen de calcitonina han sido demostrados en el líquido cefalorraquídeo (LCR) de pacientes con HSA, a partir de su liberación en las terminales nerviosas del nervio trigémino.
La producción de sustancia P, péptido relacionado con el gen de calcitonina, histamina, 5- hidroxitriptamina, endotelina 1 y bradicinina después de una HSA han sido propuestas como mediadores de disfunción de la barrera hematoencefálica, lo cual aumentaría la permeabilidad vascular y permitiría la entrada de múltiples sustancias al intersticio donde pueden ejercer efectos nocivos directos o indirectos al liberar moléculas de adhesión celular, proinflamatorios y vasoconstrictores sistémicos1,17.
3.4.2 Inflamación clásica
La sangre extravasada por una HSA sería responsable de una cascada de reacciones que llevan a la producción de varios factores vasoactivos y proinflamatorios en el espacio subaracnoideo.
Estos factores se han asociado con el desarrollo de lesiones inflamatorias de la vasculatura cerebral e incluyen:
1. La oxihemoglobina de los eritrocitos lisados.
2. Productos de la activación de la ciclooxigenasa y lipooxigenasa.
3. Endotelina 1, factor de crecimiento derivado de las plaquetas y citocinas proinflamatorias.
4. Acción protrombótica y proinflamatoria del complemento y la trombina en el endotelio.
5. Interacción de granulocitos y macrófagos perivasculares e intramurales con las moléculas de adhesión.
Se han encontrado en estudios clínicos niveles elevados de mediadores inflamatorios como IL-1 beta, factor de necrosis tumoral e IL-6 en LCR asociados con pobre pronóstico neurológico20.
Existe evidencia de que estos factores pueden desarrollar vasoespasmo en modelos animales, sin que se pueda demostrar aún lo mismo en humanos.
Es posible que en un mismo paciente se encuentren todos estos mecanismos conjugados en mayor o menor medida para originar el vasoespasmo21, 22.
4. PREDICCIÓN DEL VASOESPASMO
Infortunadamente, la predicción del desarrollo de vasoespasmo después de hemorragia subaracnoidea es imprecisa por los medios disponibles actualmente.
Los parámetros que se han postulado como factores de predicción de vasoespasmo después de HSA serán discutidos brevemente a continuación.
4.1. Volumen de sangre y presencia de HSA
La mayoría de los autores consideran que la cantidad de sangre ubicada en el espacio subaracnoideo en la tomografía cerebral inicial de pacientes con HSA, es el factor de predicción más poderoso del desarrollo de vasoespasmo1,23,24,25.
Fisher y colaboradores encontraron una alta correlación entre el volumen de sangre subaracnoidea y la aparición de vasoespasmo23. Fisher definió como factor de predicción de riesgo mayor a la presencia de coágulos de sangre de más de 5 por 3 mmen el espacio subaracnoideo medidos en la tomografía computada (Fisher 3); en un estudio publicado por su grupo, se describió que estos pacientes tenían mayores posibilidades de desarrollar vasoespasmo clínico en el territorio arterial correspondiente. En ausencia de sangrado o distribución en forma de capa delgada y difusa, el vasoespasmo sintomático ocurría en 1 de 18 pacientes23 (tabla 2).

Recientemente, Classenn y colaboradores propusieron una nueva escala modificada de Fisher26, como resultado de un estudio de 276 pacientes con HSA y tomografía cerebral en las primeras 72 horas del sangrado.
Se cuantificaron variables demográficas, clínicas y de imágenes arrojadas por la tomografía.
Los resultados de este estudio mostraron que los coágulos gruesos que llenan alguna de las cisternas basales o la cisura de Silvio, fueron el mejor factor de predicción de la aparición de isquemia cerebral tardía (p=0,008). En relación a la variable de hemorragia intraventricular, si se encontraba un sangrado que llenara ambos ventrículos laterales la correlación era todavía mayor (p=0,001)26.
Otros autores, tratando de encontrar mayor poder estadístico, han postulado medios para medir el tamaño total del coágulo y predecir el vasoespasmo; los resultados de estos trabajos junto con el de Classen no han podido ser reproducidos por otros autores24.
A pesar de las falencias ampliamente discutidas en la clasificación de Fisher como factor de predicción de vasoespasmo, es el parámetro más ampliamente aceptado para clasificar los pacientes con alto riesgo de desarrollar vasoespasmo sintomático; se incluyen en este último grupo a aquellos clasificados como Fisher III.
Además de la cantidad de sangre en el espacio subaracnoideo, el número y la gravedad de los episodios de sangrado han sido correlacionados con la incidencia de vasoespasmo. En experimentos de laboratorio se ha observado que repetidas inyecciones de sangre en el espacio subaracnoideo empeoran el vasoespasmo. Los estudios angiográficos en humanos demuestran que los episodios de resangrado inducen el desarrollo de vasoespasmo más tempranamente que un episodio único1.
4.2. Estado clínico
Otro medio que se ha utilizado como factor de predicción de vasoespasmo es el grado clínico de compromiso neurológico del paciente admitido con una HSA, con base en una de las clasificaciones propuestas para medir este aspecto.
En el estudio de Graf y Nebbelink que incluyó 274 pacientes con HSA, se encontró una clara correlación entre la evidencia angiográfica de vasoespasmo y el pobre estado clínico de ingreso. Esto significaría que la frecuencia y la gravedad del vasoespasmo se incrementaría con la gravedad del grado clínico inicial.
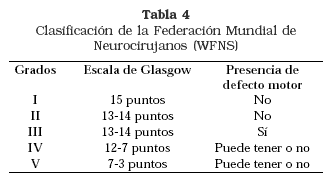
Las escalas más comunes para la evaluación del grado clínico y la predicción del pronóstico de pacientes con HSA son: la escala de Hunt y Hess, la escala de la Federación Mundial de Neurocirujanos (WFNS), la escala de Fisher y la escala de resultados clínicos de Glasgow (tablas 3, 4 y 5).


Estas escalas pueden ser imprecisas dependiendo del momento en que se apliquen durante el evento hemorrágico, lo cual influye para que los resultados de grandes cohortes con cada una de ellas reporten tasas de predicción variables, poco reproducibles y, por consiguiente, no se consideren como buenos factores de predicción de déficit neurológico tardío después de HSA por ruptura aneurismática1 (tabla 6).

4.3. Tamaño y localización del aneurisma
Un tercer factor potencial que influye en la ocurrencia de vasoespasmo es el tamaño y localización del aneurisma. La evidencia clínica de una correlación entre la localización del aneurisma roto y la incidencia del vasoespasmo no es concluyente.
Algunos reportes han sugerido que la incidencia de vasoespasmo varía con la localización del aneurisma. Por ejemplo, Fisher y colaboradores encontraron una alta incidencia de vasoespasmo después de aneurismas rotos de la arteria cerebral anterior comparada con los de la arteria cerebral media1. Grab y Nibbelink encontraron que 50% de los pacientes con aneurisma de la arteria carótida interna, 44,7% de pacientes con aneurisma de la arteria cerebral media y 35,2% de los pacientes con aneurisma de la arteria cerebral anterior presentaban vasoespasmo difuso o localizado.
Otros estudios han documentado más incidencia de vasoespasmo en aneurismas de la circulación anterior del polígono de Willis, como los de la arteria carótida interna, en relación a los de la arteria cerebral media.
McGirt y colaboradores encontraron que los pacientes con ruptura de aneurisma de la arteria cerebral posterior tenían 20 veces menos probabilidad de desarrollar vasoespasmo (P<0,005). Otros estudios encontraron que no existe relación alguna entre la localización del aneurisma y la posibilidad de desarrollar vasoespasmo1.
Al igual que la localización, el tamaño del aneurisma no es concluyente como factor de predicción de vasoespasmo. MacDonald y colaboradores analizaron datos obtenidos de 3.547 pacientes con HSA mediante una prueba de regresión logística multivariada y monovariada para determinar los factores que predecían vasoespasmo sintomático. Encontraron una correlación directa con el tamaño del aneurisma. Otros estudios no han documentado que exista tal correlación1.
4.4. Uso de cocaína
La cocaína y sus metabolitos son potentes vasoconstrictores arteriales cerebrales y causan hipoperfusión crónica cerebral tanto en modelos animales como en humanos. Podría predisponer a la ruptura del aneurisma por su efecto vasoconstrictor y facilitar el desarrollo de vasoespasmo, sin que esto se haya podido demostrar.
4.5. Sexo
Aunque las mujeres tienden a ser más susceptibles a la formación de aneurismas y a su ruptura que los hombres, no hay indicios que demuestren una mayor tendencia de ruptura de aneurismas cerebrales en mujeres que en hombres. Las series más grandes no encuentran relación del sexo con la aparición de vasoespasmo1.
4.6. Edad del paciente
Un estudio retrospectivo amplio, analizó el efecto de la edad como factor de predicción de vasoespasmo después de una HSA aneurismática.
Los pacientes fueron divididos en 2 grupos, el primero, menores de 68 años y el segundo mayores de 68 años.
Este estudio demostró que:
1. Los pacientes mayores de 68 años tienen menos incidencia de vasoespasmo (P<0,05).
2. Los pacientes de más edad tienen un promedio menor de velocidad en el Doppler transcraneal teniendo vasoespasmo clínico (P<0,003).
3. Un aumento abrupto de la velocidad de flujo en la arteria cerebral media precede el diagnóstico de vasoespasmo en dos días en ancianos y en un día en los más jóvenes.
Los investigadores concluyen que los pacientes mayores tienen una menor incidencia de vasoespasmo sintomático que los pacientes jóvenes.
Rabb y colaboradores hicieron un análisis estadístico y demostraron una predicción mayor para desarrollar vasoespasmo en pacientes menores de 35 años.
Charpentier y colaboradores demostraron que los pacientes mayores de 50 años tienen una probabilidad menor de ocurrencia de vasoespasmo.
La baja incidencia de vasoespasmo en pacientes mayores se explica por ateroesclerosis de los vasos craneales que resulta en una falla en la elasticidad y contractilidad de la pared muscular de las pequeñas arterias y arteriolas, que lleva a una respuesta incompleta de vasodilatación compensadora1.
4.7. Cigarrillo
Otro estudio prospectivo de 75 pacientes evaluó el riesgo de desarrollo de vasoespasmo por factores de predicción diferentes al grosor del coágulo de la hemorragia subaracnoidea.
En el análisis multivariado se encontró que el consumo de cigarrillo aumenta el riesgo de vasoespasmo sintomático después de una HSA aneurismática independientemente del grado de Fisher (P=0,033).
Weir y colaboradores encontraron en un estudio prospectivo de 3.500 pacientes, alta significancia (P=<0,005) de correlación entre el consumo de cigarrillo y vasoespasmo angiográfico.
4.8. Hipertensión
La hipertensión ha demostrado incrementar independiente de otros factores, el riesgo de infarto cerebral después de una HSA. El estudio de Ohman y colaboradores logró demostrar que los pacientes hipertensos presentaban infartos cerebrales en la escanografía en mayor porcentaje que los no hipertensos1.
La fisiopatología que explique lo anterior aún continúa sin esclarecerse.
5. DIAGNÓSTICO
El diagnóstico de vasoespasmo después de HSA es clínico en la mayoría de los casos y está dado por la aparición de nuevo déficit neurológico en un paciente con hemorragia subaracnoidea.
La alteración neurológica está dada por aparición de nuevo deterioro de la conciencia, afasia o déficit motor2.
Estos hallazgos pueden ser fácilmente identificados en pacientes concientes con buen estado clínico después del sangrado, pero es más dificil en aquellos con compromiso establecido de la conciencia1,5,27,28.
Tanto para los primeros, y con mayor razón para los segundos, el apoyo en los estudios neurorradiológicos son de gran utilidad para definir el riesgo de vasoespasmo, caracterizar la localización del aneurisma, organizar el plan de intervención quirúrgica y monitorizar al paciente, en aras de descartar el desarrollo de vasoespasmo o de diagnosticarlo tempranamente, todo con el objetivo de optimizar la terapia de intervención1,27.
5.1. Imágenes diagnósticas
Se acepta ampliamente que la prueba de referencia para el diagnóstico del vasoespasmo es la arteriografía cerebral9. El Doppler transcraneal es un estudio con menor sensibilidad y especificidad que la arteriografía, con importantes ventajas por ser no invasivo, de fácil acceso y menos costoso, razones por las que se propone como método principal en el seguimiento de estos pacientes9.
5.1.1 Angiografía convencional
La angiografía cerebral continúa siendo la prueba de referencia en el diagnóstico definitivo de vasoespasmo cerebral (figura 2) y todos los métodos de monitoría son comparados directamente con ella4,29.
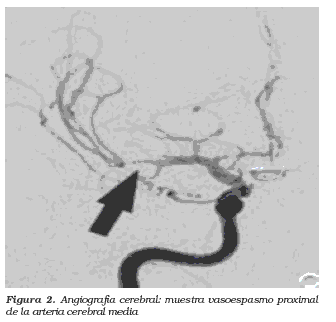
En muchos centros a nivel mundial es rutinario tomar angiografías cerebrales después de la cirugía de aseguramiento del aneurisma dentro de los primeros 7 días de evolución al sangrado inicial, para observar cambios en el diámetro de los vasos de la base del cerebro que sugieran vasoespasmo y evaluar posibles complicaciones postquirúrgicas como: oclusiones incompletas del aneurisma (5- 7%), oclusión del vaso del aneurisma (5-7%) o infartos cerebrales (2 -8%)1,30.
Se considera, en general, un método de monitorización seguro, asociado a una relativa baja morbilidad (laceraciones arteriales en 1% de los casos)30,31.
Las limitantes más importantes son: la baja pero significativa tasa de complicaciones asociadas,acceso no disponible para todos los casos y el alto costo comparado con otros métodos. Todo esto sumado al hecho de que no todos los vasoespasmos angiográficos son vasoespasmos clínicos y que el vasoespasmo confinado a vasos más distales y pequeños en el parénquima cerebral no son evaluables.
En nuestro medio, generalmente, no se utiliza la angiografía cerebral como método de evaluación de resultados postinterevención quirúrgica.
5.1.2. Doppler transcraneal (DTC)
La ultrasonografía Doppler transcraneal tiene un papel establecido para la detección y monitoreo de vasoespasmo angiográfico después de HSA aneurismática (recomendación tipo A, clase I a II)32.
En 1984, Aslid y colaboradores presentaron un trabajo en el que se encontró aumento de la velocidad del flujo en las arterias de la base del cerebro medidos por Doppler transcraneal y se correlacionó inversamente con el diámetro del vaso estudiado. Ellos proponen una velocidad media mayor de 120 cm/seg como el límite inferior para el diagnóstico de vasoespasmo33. Se consideran como valores normales de velocidad de flujo en la arteria cerebral media entre 33 y 90 cm/seg9,29.
La arteriografía, como se ha dicho, es la prueba estándar para el diagnóstico de vasoespasmo, así que comparado con ella, la mayoría de los estudios encuentra una sensibilidad entre el 60% y el 100% y una especificidad del 80% al 100%1,34,35; sin embargo, es un método no apropiado para hacer mediciones seriadas, mientras que el DTC nos permite hacer mediciones a repetición sin la necesidad de trasladar al paciente34.
En una revisión sistemática reciente con recopilación de los mejores estudios disponibles, se compara el Doppler transcraneal con la angiografía cerebral, se encontró que para vasoespasmo de la arteria cerebral media (ACM) la sensibilidad del Doppler transcraneal comparada con aquélla es de 67% (95% IC 48% - 87%), especificidad de 99% (98% - 100%), valor predictivo positivo (VPP) de 97% (95% - 98%) y valor predictivo negativo (VPN) de 78% (65% - 91%).
Para la arteria cerebral anterior, se reportó una sensibilidad de 42% (11% - 72%), especificidad de 76% (53% a 100%), VPP de 56% (27% a 84%) y VPN de 69% (43% a 95%).
Esto implica que resulta un método útil y recomendado para monitoría de la arteria cerebral media, pero no para el seguimiento de la arteria cerebral anterior. Los autores, además, concluyen que no existe evidencia suficiente para recomendar su uso como monitoreo de la circulación posterior9.
Suárez y colaboradores encontraron que cuando se evalúan los resultados con relación al desarrollo de vasoespasmo sintomático, el VPP es de 39% y el VPN de 90%36; considera que velocidades de flujo por encima de 120 cm/seg se asocian con leve a moderado vasoespasmo, velocidades por encima de 200 cm/seg están asociadas con vasoespasmo grave37,38.
Vora y colaboradores sugieren que sólo con velocidades muy altas o muy bajas en la arteria cerebral media (< de 120 cm/seg o > de 200 cm/seg) se predice en forma positiva (87%) o negativa (94%) el vasoespasmo sintomático, mientras que valores intermedios tienen un valor predictivo bajo31.
La mayoría de los autores coinciden en considerar como el límite inferior para hablar de vasoespasmo una velocidad sistólica pico de la arteria cerebral media de 120 cm/seg39.
Alternativamente, se ha postulado que mediciones seriadas mejoran la calidad de los datos mostrados por la prueba; con base en lo anterior se considera que aumentos mayores de 25 - 65 cm/seg en 24 horas de seguimiento son buenos indicadores de inicio de vasoespasmo7,19,38,39.
En forma indirecta se puede hallar el índice de Lindegaard, definido como la relación entre la velocidad de flujo en la arteria cerebral media y la que se encuentre en la arteria carótida interna ipsilateral, si este índice es mayor 3 indica la presencia de vasoespasmo y si es mayor de 6, lo clasifica como grave31,34.
Estudios retrospectivos han comparado las pruebas de medición de perfusión con el DTC demostrando que las velocidades altas podrían ocasionalmente corresponder a un flujo compensatorio y no necesariamente a un vasoespasmo.
La limitante más importante con este estudio es la dependencia del operador, el porcentaje de pacientes en los que resulta imposible encontrar el flujo del vaso sanguíneo a estudiar (puede llegar al 10% de los casos)4, el porcentaje importante de falsos negativos en pacientes que teniendo compromiso del flujo sanguíneo cerebral tienen trastornos de autorregulación y desarrollan vasodilatación distal al vaso comprometido, esto puede impedir un aumento relevante de las velocidades de flujo medidas9, además, todo trastorno del tono de vasos arteriales distales escapa al alcance de la evaluación de este método imaginológico, aunque muchos autores consideran que esto último es infrecuente y tiene poca importancia en la práctica clínica40.
La recomendación de mayor aceptación en cuanto al momento de iniciar la monitorización y la frecuencia de la misma es que en pacientes de alto riesgo se inicie desde el primer día de sangrado y luego diariamente si fuera posible o cada dos días41 hasta los días 7 al 10 si el paciente está asintomático o, por el contrario, si es sintomático su utilidad se prolongaría hasta los días 12-18 del sangrado inicial33,34. Por supuesto que si el paciente desarrolla signos agudos sugestivos de vasoespasmo cerebral, como son nueva aparición de afasia, déficit motor o compromiso de la conciencia, ameritaría otra evaluación con este método inmediatamente.
Una propuesta de seguimiento sería realizar un DTC el día de ingreso para tomar valores basales y continuar los días 3, 5, 7, 9, 11 y 13 si no hay cambios clínicos sugestivos o inmediatamente después de la presencia de un nuevo hallazgo clínico.figura 3

5.1.3. Tomografía cerebral simple, imagen de perfusión por escanografía cerebral o resonancia magnética nuclear
La tomografía cerebral simple se utiliza como estudio inicial del paciente con HSA, tanto para al diagnóstico como para la predicción del vasoespasmo en estos pacientes23,26. La cantidad de sangre en el espacio subaracnoideo es factor de predicción de infartos producidos por 15 vasoespasmos. Así mismo, este método permite evaluar con aceptable calidad el desarrollo de complicaciones concomitantes al vasoespasmo, como resangrado y aparición de nuevas áreas de isquemia.
La escanografía de perfusión ha demostrado ser una buena alternativa para evaluar el flujo sanguíneo cerebral1. Además, es un método de amplia disponibilidad y menor complejidad que otros, como la resonancia magnética.
La escanografía de perfusión contrastada con xenón ayuda a distinguir entre la isquemia reversible y el infarto irreversible. El límite considerado de vasoespasmo por este método es de 20 centímetros cúbicos de sangre por cada 100 gramos de tejido.
Las desventajas de este estudio son: radiación del cráneo, alteraciones en el estado mental y efectos secundarios del xenón en la perfusión cerebral.
Estas técnicas son cada vez más utilizadas a nivel mundial para el diagnóstico temprano de déficit neurológico tardío y seguimiento.
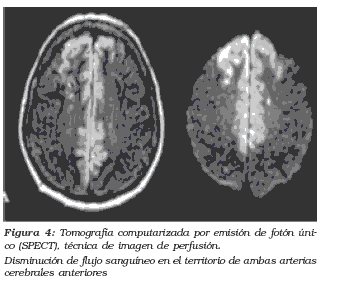
La tomografía computarizada por emisión de fotones (SPECT), se haya bien establecida en la valoración del flujo sanguíneo cerebral. A diferencia del DTC, el SPECT puede valorar la perfusión cerebral a nivel celular. Es utilizado en pacientes con pobre estado neurológico que no pueden ser evaluados adecuadamente por deterioro clÃnico. Proporciona sensibilidad (89%) y especificidad (75%) para la detección del vasoespasmo1.figura 4
6. EXCLUSIÓN DEL ANEURISMA Y TIEMPO DE TRATAMIENTO
El aseguramiento del aneurisma por medio quirúrgico con clipaje directo o por oclusión con coils de colocación endovascular es parte fundamental del manejo del paciente con HSA. Ambos métodos son plenamente válidos y, aunque existe controversia, los estudios diseñados para evaluar diferencias no han encontrado que éstas son significantes42, 43,44 o describieron ventajas y desventajas en grupos específicos45.
Existe actualmente una tendencia cada vez mayor a hacer más procedimientos de exclusión de aneurismas por técnicas endovasculares46.
Varios estudios han demostrado que los aneurismas localizados en la circulación posterior, sobre todo aquellos ubicados en la punta de la arteria basilar, tienen mejor resultado con terapia endovascular43, mientras que los ubicados en la bifurcación de la arteria cerebral media son mejor tratados con clipaje directo.
El manejo con terapia endovascular tiene la ventaja de poder ser realizado en cualquier momento de la evolución del paciente (aun en el período de mayor riesgo para vasoespasmo), con efectividad aun en pacientes de alto grado clínico (Hunt y Hess mayor de 2)46.
Igual controversia existe con respecto al momento de intervención del paciente para clipaje definitivo, aunque un buen número de trabajos muestran que es ventajosa la intervención temprana porque reduce la tasa de resangrado4,47,48, no han podido demostrar que esto resulte en una mejoría global del paciente en cuanto a morbilidad y mortalidad; sin embargo, la tendencia es la intervención temprana antes del tercer día46,49. Lo que sí es claro es que la mayoría de los estudios han mostrado que del día 3 al 14 claramente la cirugía se asocia a la mayor morbilidad por el pico de vasoespasmo que se observan en estos días46.
Algunos estudios han mostrado pequeñas ventajas para la terapia de intervención temprana o intermedia en pacientes con grados clínicos bajos (Hunt and Hess menor de 3)46,49.
Las ventajas potenciales de la intervención temprana son evidentes: evitar el resangrado y permitir la instalación agresiva de la terapia triple H46,49.
Sin embargo, la decisión de si el paciente se va a intervenir, el medio que se va a utilizar y el momento de hacerlo dependen en primera instancia de la evaluación inicial del clínico que valore al paciente con HSA aneurismática.
7. ESTRATIFICACIÓN DEL RIESGO DE DESARROLLO DE VASOESPASMO SINTOMÁTICO
Se ha descrito una serie de factores de riesgo para predecir qué pacientes tienen mayor riesgo de presentar esta complicación y requerir una terapia más agresiva.
De todos ellos, el más consistente para anticipar un episodio de vasoespasmo es la presencia de sangrado masivo en las cisternas de la base o el llenado de ambos ventrículos laterales1,5 así mismo, aquellos pacientes que en el seguimiento de Doppler transcraneal presentan una elevación continuada de las velocidades de flujo sanguíneo o aumento en la velocidad sistólica pico medida en 24 horas de 25 - 65 cm/seg entrarían en el protocolo de tratamiento agresivo como pacientes de alto grado de sospecha de vasoespasmo cerebral29,33,34. Los dos anteriores parámetros deberían ser tomados como medio de estratificación y con uno solo de ellos el paciente pasaría al grupo de alto riesgo para desarrollo de vasoespasmo sintomático.
Otros factores de riesgo han sido mencionados como factores de predicción: hipovolemia, hiponatremia, tabaquismo, adicción a cocaína, aneurismas grandes por arteriografía, aneurismas de la arteria cerebral anterior, sexo femenino y pacientes menores de 55 años1,5.
8. TRATAMIENTO DEL VASOESPASMO
El manejo del vasoespasmo después de hemorragia subaracnoidea (HSA) cada vez apunta más a terapias dirigidas a evitar su presentación (tratamiento profiláctico), diagnóstico hiperagudo e intervención enérgica cuando se ha desarrollado, ya que sigue siendo la principal causa de morbilidad y mortalidad en supervivientes de HSA1,2,4,5,6.
El tratamiento del vasoespasmo se divide en las siguientes categorías mayores:
- Tratamiento médico
- Tratamiento endovascular
El tratamiento se inicia con la prevención del vasoespasmo, utilizando medidas que favorecen la normo o hipervolemia, la hipertensión, la hemodilución y el uso de calcio-antagonistas que pudieran disminuir la posibilidad de presentación del mismo y con ello de las graves complicaciones asociadas1, 2,4,27,50.
8.1. Tratamiento médico
Incluye la instauración de la terapia triple H y el uso de calcioantagonistas como principales medidas.
8.1.1 Terapia triple H
(hipervolemia, hipertensión y hemodilución)
Se trata de la inducción de hipervolemia, hipertensión y hemodilución como medidas que aumenten el flujo sanguíneo cerebral tanto en forma profiláctica como terapéutica para vasoespasmo cerebral4,6,27,51, con un nivel de evidencia III a V, grado C4; esto implica que el peso de la evidencia que respalda su uso en la práctica clínica no es muy grande; sin embargo, es ampliamente utilizado a nivel mundial y una serie de estudios de pequeño tamaño ha mostrado su utilidad práctica. Actualmente a,lgunos autores llaman a esta intervención terapia de 4 H por la inclusión voluntaria o no de hiperdinamia.
La terapia se instaura en forma escalonada, esto depende del momento de llegada del paciente en relación a la fecha de sangrado inicial, clasificación de riesgo de desarrollar vasoespasmo (según clasificación de Fisher) y si el aneurisma ha sido o no asegurado.
Lo anterior es importante para decidir la intensidad de la terapia triple H.
El método consiste en lograr metas específicas para cada uno de estos parámetros de acuerdo con el momento de evolución, teniendo en cuenta que, según lo anterior, podrá ser más o menos agresiva durante la evolución de cada paciente y, por tanto, esta decisión se basa en la evaluación clínica y paraclínica de cada momento del tratamiento; la implementación completa de todas estas metas en todos los pacientes podría traer riesgos importantes en algunos ellos (por ejemplo, riesgo de resangrado en aquéllos con aneurismas no asegurados).
En la actualidad, esta terapia se considera benéfica y es ampliamente utilizada en clínica a pesar de no tener soporte de evidencia suficiente4,6,50,51.
El siguiente orden es el recomendado para la instauración escalonada de la terapia, pasando de la menor intervención a la mayor, si no hay mejoría rápida de los síntomas.
8.1.1.1 En todos los pacientes, previo al aseguramiento del aneurisma, se conservará normovolemia y normonatremia y el primer parámetro deberá ser monitorizado con un catéter venoso central, manteniendo una presión venosa central (PVC) entre 8 y 10 cm de H20. Se mantendrá la PAM en valores cercanos a 100 o 110 mm Hg, dado que la mayoría de los pacientes por autorregulación ingresan con tensiones arteriales cercanas a este nivel para mantener la presión de perfusión cerebral; si el paciente se encuentra hipotenso, ingresará a un protocolo de normalización de la tensión arterial con líquidos endovenosos, inotrópicos o vasoconstrictores de ser el caso, para evitar el riesgo de isquemia cerebral.
En pacientes con hipertensión severa (TAM>140 mm Hg) se utilizarán betabloqueadores si no están contraindicados o vasodilatadores sistémicos, en ese orden, con el fin de disminuir la posibilidad de resangrado inmediato. Se tendrá como ideal una hemodilución con hematocrito entre 30 y 40%; sin embargo, esta no se forzará y en estos casos se logrará sólo con la administración de líquidos endovenosos necesarios para mantener normovolemia5,27.
8.1.1.2 Una vez que el aneurisma ha sido asegurado con clips o coils de colocación endovascular se procederá a incluir al paciente según factores de riesgo en el grupo de alto o bajo riesgo para continuar la terapia.
8.1.1.2.1. Pacientes de bajo riesgo y aneurisma asegurado: monitoría con catéter venoso central, línea arterial, mantener normovolemia con PVC de 8 y10 cm/H20, TAM alrededor de 110 mm Hg, hemodilución con hematocrito entre 30 y 40% instaurada en el transcurso de los siguientes 3 días como mínimo5,27.
8.1.1.2.2. Pacientes de alto riesgo y aneurisma asegurado: los pacientes de alto riesgo recibirán al máximo la terapia triple H nuevamente de forma escalonada. En el postoperatorio inmediato se colocará un catéter de arteria pulmonar dado que una agresiva hipervolemia puede llevar a complicaciones asociadas a la terapia (del 3,5 a 17% de desarrollo de edema pulmonar sintomático)5,11, 27 más en aquellos pacientes con comorbilidades como disfunción cardíaca o pulmonar, se mantendrán PVC entre 10 a 12 y presión de oclusión en la arteria pulmonar entre 12 y 14, se instalará una línea arterial para mantener TAS entre 150 y 170 mm Hg.
8.1.1.3 En pacientes en los que se ha comprobado vasoespasmo clínicamente o por arteriografía cerebral o Doppler transcraneal, se recomienda llevar la presión de oclusión de la arteria pulmonar estrictamente a niveles superiores a 14 cm H20; de no lograrse mejoría clínica en una hora27 se llevará la tensión arterial sistólica a 180 mm Hg o incluso 200 mm Hg en sintomáticos, lo que generalmente necesitará de vasopresores; algunos autores prefieren iniciar con fenilefrina en dosis de 10-30 µg/min de infusión continua y sólo con noradrenalina (0,05 - 0,2 µg/kg/m) cuando la infusión de fenilefrina no logra los objetivos deseados.
Por último, si no se consiguen los objetivos establecidos en una hora, se procederá al uso de inotrópicos como dobutamina, dopamina o milrinone para aumentar el gasto cardíaco52 y, con ello, el flujo sanguíneo en las áreas cerebrales comprometidas, siendo el objetivo un índice cardiaco mayor de 3,5 l/min/m2 27. En caso que la terapia con calcioantagonistas sistémicos y la terapia triple H sean insuficientes para mejorar clínicamente al paciente, se procederá a instaurar otros métodos de manejo como la angioplastia percutánea temprana del vaso comprometido, idealmente dentro de las primeras 2 horas de evolución o, al menos, dentro de las primeras 24 horas de vasoespasmo7,8,27,20, 53, 54,55.
Habitualmente se prefieren los coloides o la mezcla de coloides y cristaloides para lograr la meta de hipervolemia en terapia triple H, por la mayor facilidad para lograr dichas metas en corto tiempo.
Con mucha frecuencia es difícil mantener hipervolemia e hipertensión dada la natriuresis y, por tanto, la poliuria que presentan los pacientes en terapia triple H con función renal normal. Varios medicamentos se han utilizado para lograr estos objetivos. La vasopresina ejerce efectos sobre el volumen sanguíneo total al favorecer la retención de agua libre en el túbulo contorneado distal y túbulo colector de la nefrona. Al administrarse a pacientes sometidos a terapia triple H, favorece el mantenimiento del estado hipervolémico al reducir la diuresis. Las dosis usualmente utilizadas van entre 1 y 4 UI cada 6 a 8 horas, ajustando la dosis según necesidad por vía subcutánea. Un aspecto importante del uso de vasopresina es que dado su mecanismo de acción, es necesario mantener un estricto control de la natremia, por el riesgo de hiponatremia al usar este medicamento y mantener control estricto de los signos vitales, por la posibilidad de una respuesta hipertensiva importante56.
Otro método utilizado para evitar las pérdidas urinarias altas es la administración de fludrocortisona a dosis de 0,1 - 0,3 mg cada 12 horas por vía oral27; en reportes recientes se documenta el uso de hidrocortisona, 100 mg cada 8 horas endovenosa, que tendría las ventajas teóricas de tener una vida media más corta y ser, por tanto, más titulable y con efectos adversos menos serios, su uso no se acompañó de complicaciones como aumento de infecciones o sangrado gastrointestinal11.
Recomendamos la utilización de vasopresina, sin olvidar la necesidad de monitoreo estricto de los niveles de sodio sérico.
Un aparte que no se debe dejar de lado es la posibilidad que un paciente presente a la vez más de un aneurisma, en cuyo caso estaríamos ante el dilema de provocar la ruptura del aneurisma no roto; la recomendación basada en una serie de casos amplia es instalar la terapia triple H sin restricciones dado que la posibilidad de ruptura del otro aneurisma es muy baja durante este tratamiento28.
Las contraindicaciones más comunes para el uso de terapia triple H son: edema cerebral, infarto cerebral establecido, edema pulmonar de cualquier etiología, anemias moderadas o graves, síndrome de dificultad respiratoria agudo e hipertensión endocraneana (HEC).
Las complicaciones más frecuentes son el edema pulmonar, extensión del infarto cerebral o edema cerebral por hipervolemia.
8.1.2 Calcioantagonistas
La única terapia que ha demostrado claramente disminuir la posibilidad de eventos adversos desfavorables por vasoespasmo, como son muerte, estado vegetativo o discapacidad grave, es el uso de nimodipina a dosis de 60 miligramos cada 4 horas por vía oral o por sonda nasogástrica por un período de 21 días, desde el inicio de la hemorragia subaracnoidea57; de ser utilizada por vía endovenosa, la dosis varía de 0,5 a 2 mg/h; esta última vía de administración no logró demostrar el mismo grado de efectividad, por la mayor asociación de hipotensión cuando se utilizó.
En una revisión sistemática de la literatura reciente mostró que la terapia con calcioantagonistas redujo el riesgo de resultado desfavorable (déficit neurológico tardío o infarto cerebral confirmado por imágenes) con un riesgo relativo (RR) de 0,82, intervalo de confianza (IC) del 95% (0,72 - 0,93), reducción del riego absoluto de 5,1%, y un número necesario a tratar (NNT) de 20. Cuando se utilizó nimodipina oral como único calcioantagonista el RR de eventos adversos fue de 0,7 (0,58-0,84). Para todos los calcioantagonistas el RR de muerte fue 0,9 (IC de 95%: 0,76 - 1,07). Para nimodipina el RR de déficit neurológico tardío 0,65 (IC de 95%: 0,51 - 0,82), con un NNT de 8 y el de infarto confirmado por imágenes de 0,7 (IC de 95%: 0,58 - 0,85), con un NNT de 757,58.
Esta terapia se recomienda como una intervención preventiva en todos los pacientes con HSA sin importar la clasificación o si pertenece o no al grupo de alto riesgo para vasoespasmo.
El evento adverso más frecuente con nimodipina fue hipotensión; se encontró en 2,1% en el grupo de tratamiento contra 1,4% en el grupo control57.
Como se mencionó antes, la revisión sistemática de la colaboración Cochrane encuentra mejor el uso de nimodipina por vía oral que por vía endovenosa por el desarrollo más frecuente de hipotensión cuando se utiliza esta última; por ello, el seguimiento y probablemente la necesidad de inotrópicos y vasopresores para mantener las metas hemodinámicas debe ser más estricto al utilizar esta vía57,58.
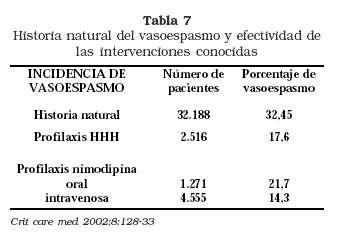
La tabla 7 muestra los resultados de una recopilación de estudios de pacientes con hemorragia subaracnoidea y el resultado de la implantación de tratamiento médico con terapia triple H y calcioantagonistas en cuanto a prevención de aparición de vasoespasmo y el resultado final del mismo.
8.1.3 Terapia con antifibrinolíticos
Es altamente controversial el uso de estos fármacos en este grupo de pacientes. El objetivo de su administración se centra en la capacidad de bloquear la unión de la plasmina a la fibrina del coágulo sanguíneo y con ello tener la esperanza de reducir el riesgo de resangrado. En estudios previos se ha encontrado que si bien estos medicamentos (ácido epsilon-amino caproico y ácido tranexámico) disminuyen el resangrado temprano después de HSA, es claro que aumentan la posibilidad de isquemia cerebral cuando se administran en forma prolongada, por lo que no se ha encontrado un efecto beneficioso claro59. Por ello, aunque en algunos centros se utiliza en forma temprana hasta el aseguramiento del aneurisma roto12,60, no recomendamos su uso sistemático para el tratamiento de vasoespasmo posterior a ruptura de aneurisma cerebral.
8.1.4 Drenaje de líquido cefalorraquídeo Se ha reportado que el drenaje de líquido cefalorraquídeo en pacientes con HSA reduce la aparición de vasoespasmo, además, mejora el resultado y la necesidad de intervención en los pacientes que desarrollan esta complicación61,62. El uso de este método de tratamiento es generalmente una decisión basada en el beneficio esperado en cada paciente después de individualizar el riego/beneficio para éste.
8.2 Terapia endovascular La gran mayoría de la literatura actual, con algunas excepciones53, apoya el uso de terapia endovascular para el tratamiento de pacientes que estén recibiendo terapia de prevención desarrollan vasoespasmo clínico y éste no mejora a pesar del establecimiento de terapia médica máxima (hasta el 15% de supervivientes de un primer sangrado por HSA)5,11,18,27,51,54,63,64,65.
La tendencia es cada vez mayor a hacer intervenciones más tempranas (dentro de las primeras 24 horas); en este aspecto, algunos abogan por una ventana de intervención de 2 horas desde el inicio de vasoespasmo clínico (mejoría de 70% en tratados antes de 2 horas y de 40% en tratados después de este lapso)11 o, incluso, se ha utilizado de forma profiláctica en pacientes de alto riesgo7.
Para llevar al paciente a un tratamiento de terapia endovascular es necesario estar seguro del diagnóstico de vasoespasmo al encontrar un deterioro nuevo en el estado neurológico del paciente particularmente, entre el día 3 y 14 de la primera hemorragia o hallazgos en el monitoreo de doppler transcraneal compatibles con ello, como una velocidad de flujo en la arteria cerebral media mayor de 120 cm/seg (máximo poder de especificidad con velocidades mayores a 200 cm/seg), índice de Lindegaard mayor de 3 o aumento en la velocidad de flujo en ACM mayor de 50 cm/seg entre una medida y la siguiente7. Algunos autores incluyen la presencia de fiebre como indicador de presencia de vasoespasmo y de mal pronóstico en general, independiente o no de si los pacientes presentaban infección manifiesta66.
Todos los pacientes deben tener un estudio de tomografía computarizada reciente que excluya la posibilidad de otras causas cerebrales que expliquen el deterioro neurológico del paciente como áreas de infarto, isquemia o hidrocefalia que podrían contraindicar el procedimiento. Se recomienda que el paciente se encuentre anestesiado y con relajación muscular, sobre todo, si se va a realizar angioplastia8.
Se han reportado resultados favorables con la terapia endovascular, con reducción de hasta el 15% de mortalidad intrahospitalaria67 y se proponen algunos indicadores de mal resultado a pesar de la instauración de tratamiento endovascular como son clasificación pobre del estado clínico (WFNS IV o V), edad avanzada o compromiso de múltiples territorios vasculares68,69.
Para aquellos pacientes que tienen vasoespasmo al momento del ingreso hay series que proponen el tratamiento endovascular para la oclusión del aneurisma junto con terapia endovascular para el vasoespasmo, ya sea por angioplastia primaria o farmacológica con papaverina, con resultados bastante favorables teniendo en cuenta el alto riesgo de este grupo particular de pacientes64.
La terapia endovascular para vasoespasmo cerebral se puede proveer de dos formas: terapia mecánica, terapia química con administración de medicamentos vasoactivos o calcioantagonistas que favorecen la vasodilatación del segmento comprometido o del lecho distal al vaso estrechado.
8.2.1. Terapia endovascular mecánica
Consiste en angioplastia directa de las arterias comprometidas (sólo deben incluirse las arterias proximales de la base cerebral) con catéteres endovasculares que tienen características particulares; el objetivo terapéutico se consigue puesto que la dilatación vascular revierte la aumentada velocidad de circulación de la sangre a través de los vasos comprometidos, mejorando así la perfusión18, 70. El mecanismo íntimo por el que mejora el flujo vascular no es totalmente claro; en estudios experimentales en animales y humanos usando técnicas de microscopía electrónica, se encontró que la lámina elástica interna vascular se encuentra arrugada con proliferación del tejido conectivo y del músculo liso, ello conlleva a estrechamiento físico de la luz; la angioplastia mostró quitar las arrugas de la lámina elástica y estrechar el espacio ocupado por el tejido conectivo y muscular engrosado, con mínima o ninguna lesión endotelial, lo que se tradujo en dilatación efectiva del vaso comprometido18,19.tabla 8

Los materiales disponibles para el tratamiento endovascular son catéteres con balón distal dirigidos por flujo o bien catéteres sobre guía que fueron introducidos en la práctica clínica en forma más reciente. La característica más importante de estos dispositivos es que el material del balón distal es silicona y que están especialmente diseñados para aceptar altos volúmenes y crear bajas presiones de contacto con el vaso a dilatar y con ello reducir la posibilidad de ruptura que es la complicación más frecuente del procedimiento y la de mayor morbilidad8,19.
Para este procedimiento se recomienda anticoagulación plena con heparina no fraccionada para obtener tiempos de coagulación activados de dos a tres veces el basal como principio terapéutico.
El objetivo es hacer angioplastia a los vasos estrechos que se estén correlacionando con el deterioro clínico y aquéllos que en comparación con el estudio basal angiográfico, tengan una estrechez mayor de 50% siempre y cuando sea técnicamente posible abordarlos8.
Las mayores dificultades para lograr el paso del catéter se encuentran con las arterias cerebrales posteriores y cerebral anterior (menos de 10% de éxito); estos segmentos arteriales podrían ser tratados con agentes químicos.
La recomendación es la dilatación del vaso hasta un 25% de la luz normal y, luego, se continúa inflando muy suave y lentamente hasta que llegue a la apariencia previa al espasmo para, entonces desinflar rápidamente el balón8.
Está descrita una tasa de éxito hasta de 55% a100% para mejoría inmediata del vasoespasmo angiográfico con angioplastia utilizando balón o combinada con medicamentos, comparada con el 40% encontrado en pacientes con angioplastia química11,54,65,71,72, con tasas de mejoría clínica que oscilan entre 50% y 76% y de 35% a 50%, respectivamente11,54,65,67,70,71,73; como dijimos, se ha descrito que influyen en el resultado del tratamiento en forma adversa la edad avanzada y el estado clínico del paciente al ingreso al hospital68,71. La falla clínica se encuentra entre 16% y 69% de los casos8.
Se reportó alto índice de prevención de vasoespasmo en algunas series con tratamiento profiláctico con angioplastia mecánica aunque con riesgos altos de ruptura vascular55. La mortalidad con este procedimiento es de 2-5% y la morbilidad de 4-8% en series relativamente pequeñas de casos41, 54,74.
La evolución a largo plazo de pacientes con angioplastia no está disponible. Se citan como desventajas de este método la ruptura vascular, desplazamiento del clip, oclusión completa del vaso comprometido o resangrado de aneurismas no “clipados”65, lo que hace necesario que sea realizado por personal experto y suficiente donde se disponga de una adecuada cantidad de tiempo7,8.
Una reciente revisión de artículos publicados, en los que se utilizó terapia endovascular para vasoespasmo con angioplastia mecánica, reunió un total de 530 pacientes, de los cuales, 62% mejoró clínicamente, con una mortalidad asociada de 5% y 1,1% de ruptura vascular18. Se estudió en un subgrupo de pacientes las velocidades de flujo arterial medidas con doppler transcraneal, se reportó mejoría de 69%. En otro subgrupo en el que se evaluó el flujo sanguíneo regional medido por tomografía con Xe radiactivo o SPECT, se encontró mejoría de 85%18.
8.2.2. Terapia química
Hasta este momento se ha descrito una amplia variedad de medicamentos para aplicación directa intraarterial en pacientes con vasoespasmo; de todos, la mayor experiencia se tiene con la aplicación de papaverina, un potente vasodilatador que produce mejoría angiográfica de la lesión en 67 a 98% de los casos y mejoría clínica muy dispar y variable entre 0 y 100%18,41,69,75.
La papaverina es un alcaloide derivado del opio y, al parecer, el efecto es mediado por inhibición de la actividad local de la fosfodiesterasa del AMPc y GMPc18,75. En estudios experimentales se encontró que este medicamento produce efectos más notorios cuando se administra en las etapas tempranas del sangrado cuando hay pocos cambios morfológicos vasculares.tabla 9

Generalmente, se coloca en dosis que no superen los 500 miligramos por día; la dosis habitual de administración es de 200 a 300 mg por vaso comprometido, a concentraciones no mayores de 0,8%, en infusión muy lenta, lo que permite los efectos deseados en el área comprometida y evita lesión directa del vaso.
Los efectos colaterales encontrados con la medicación son: déficit focal transitorio, elevaciones de la presión intracerebral (PIC), dilatación pupilar o convulsiones8,69. La elevación de la PIC ha sido el evento adverso más frecuente y potencialmente grave descrito en una serie con un número representativo de casos, en el que se encontró elevación significante de la PIC en 42% (mayor a 20 mm Hg), de éstos, el 10% falleció por esta causa y en los supervivientes se asoció como factor de mal pronóstico, definido por la escala GOS69. La administración en el territorio de la arteria cerebral posterior se asocia a riesgo de paro respiratorio o disfunción cardíaca8,69.
Las principales ventajas de la papaverina en terapia endovascular, así como la de otros agentes endovasculares, son:
1. Alcanza segmentos arteriales de difícil acceso para angioplastia con balón, como el segmento A1 de la arteria cerebral anterior.
2. Menor riesgo teórico de ruptura vascular.
3. Parece ser más seguro intervenir pacientes con vasoespasmo sintomático y aneurisma no “clipado”, por la posibilidad de tratar únicamente los segmentos distales al aneurisma roto.
4. Parece ser más segura para tratar vasoespasmos que ya tengan áreas asociadas de hipodensidad en el TC de cráneo.
La corta vida media (aproximadamente, una hora) hace necesario reintervenir en forma frecuente a los pacientes, guiados por el estado clínico o el resultado de estudios imaginológicos11,18,27,69; otros factores, como la presencia de alta cantidad de sangre en el espacio subaracnoideo al momento del diagnóstico61 y lesión de reperfusión11, son causas probables para explicar la poca efectividad de la droga, reflejado en la disminución del entusiasmo previo con su uso67,69,75; además se recomienda instituir monitoría de la PIC cuando se utiliza en infusión intraarterial y limitar su uso a pacientes que tienen vasoespasmo de un territorio arterial único18,41,69.
Una serie reciente revisó un número grande de pacientes reunidos de varios estudios. Se encontró un total de 346 pacientes tratados con papaverina para vasoespasmo cerebral, con mejoría clínica de 43%; la necesidad de repetir el procedimiento por recaída clínica fue de 1,7 veces por paciente; a 51 se les evaluó mejoría de flujo doppler transcraneal, que mejoró en 57% de ellos aunque no por más de 48 horas18.
Otra serie de medicamentos, como calcioantagonistas e inhibidores de proteincinasa, se han utilizado por vía endovascular en el tratamiento de vasoespasmo, entre ellos, el nimodipino intraarterial parece ser promisorio; este medicamento bloquea la entrada de calcio a la célula del músculo liso vascular a través de canales lentos de calcio voltaje dependientes; de esta manera, produce vasodilatación sin alterar la actividad metabólica cerebral, y al disminuir la carga de calcio intracelular en las neuronas isquémicas puede tener efecto protector cerebral63.
El nimodipino se utiliza en infusiones cortas de 20-30 minutos a razón de 0,1 mg/min, de una solución previamente disuelta en solución salina y cuya concentración sea de 50 µg/ml, esta dosis se debe administrar a cada vaso comprometido, queda por esclarecer cuánto tiempo debe pasar entre una dosis y la subsiguiente76 tabla 10

Habitualmente el paciente recibe de 3 -4 mg por vaso tratado y la dosis total dependerá en gran medida del número de vasos afectados63.
La instauración de esta terapia no excluye el uso continuado de nimodipina por vía oral, siempre que se mantengan las metas de tensión arterial descritas previamente.
En un estudio reciente se documentó el uso de esta medicación en 30 pacientes con vasoespasmo de menos de 24 horas de evolución con el esquema previamente descrito, 19 de ellos (76%) tuvieron mejoría clínica; de éstos, sólo 12 (63%) mejoría angiográfica; después de 12 meses de seguimiento a un total de 18 pacientes (67%), se les encontró resultado favorable medido por escala de Rankin modificada76.
Los efectos adversos más frecuentemente observados con esta terapia son hipotensión, bradicardia y menos frecuentemente exantema y diarrea63,76.
La nicardipina es otro de los medicamentos utilizados para este propósito, la dosis varía entre 0,5- 0,6 mg por vaso comprometido, teniendo resultados favorables por mejoría angiográfica, y medida por DTC hasta en el 100% de los casos por menos de 4 días, la mejoría neurológica se describe en 42%.
Al igual que otros fármacos, se asocia con la elevación transitoria de la PIC en 30 a 35% de los pacientes18,77. La presentación parenteral no está disponible en nuestro país.
El verapamilo también ha sido estudiado en este grupo de pacientes, las dosis utilizadas están alrededor de 40 µg/kg por vaso comprometido. La mejoría neurológica reportada es de 20%. Los efectos colaterales son mínimos en el estado hemodinámico y en la elevación de la PIC78.
Se recomienda a los pacientes que van a terapia endovascular como tratamiento de vasoespasmo, recibir anestesia general y relajación neuromuscular, con el fin de evitar movimientos bruscos que permitan la ruptura intraoperatoria del vaso intervenido8.
8.3 Nuevas terapias
El grupo de fármacos utilizados tanto en experimentos de laboratorio como en series de casos pequeñas en humanos es amplio, citaremos algunas de ellas de acuerdo con el grado de relevancia que tengan los resultados clínicos arrojados.
La terapia cisternal de administración de activador tisular del plasminógeno recombinante (rTPA) elimina los coágulos formados en el espacio subaracnoideo después de HSA.
Ha mostrado resultados contradictorios para protección contra vasoespasmo cerebral; el estudio más grande hecho en 100 pacientes con HSA en el que se aplicó rTPA directamente en la cisterna magna durante el procedimiento de “clipaje” del aneurisma, demostró similar incidencia de vasoespasmo angiográfico para ambos grupos (control/tratados), aunque en este último grupo el desarrollo de vasoespasmo fue menos grave79.
El tirilazad, un medicamento del grupo de los 21 aminoesteroides, con una gran capacidad barredora de radicales libres ha sido estudiado en varios ensayos clínicos. En uno de ellos se encontró que no había modificación en la mortalidad global o en el desarrollo de déficit neurológico tardío, pero cuando se estudiaron sólo los varones con puntuación en la escala de Hunt y Hess de IV y V, se encontró una disminución de la mortalidad de 33% sin tratamiento a 5% en el grupo tratado80.
En otro estudio, la dosis de 15 mg/kg/día se relacionó con una disminución significativa, y al realizar una nueva estratificación por grupos, la disminución de la mortalidad en pacientes con grado neurológico IV a V de la escala de Hunt y Hess fue notoria81.
Estas intervenciones y otras como el nitroprusiato de sodio, fasulid, milrinone o inhibidores de la endotelina I (clazosentan)82 se muestran prometedoras como terapias que disminuyen la incidencia y mejoran el pronóstico en este grupo de pacientes; sin embargo, hasta ahora no se ha podido demostrar que alguno de ellos tenga suficiente evidencia clínica para ser utilizados en todos los casos de vasoespasmo por hemorragia subaracnoidea aneurismática.
8.5 Tratamientos adyuvantes
Las convulsiones asociadas a HSA y vasoespasmo no son infrecuentes (5 hasta 25% de los casos), su presencia en pacientes que han sangrado y aún no se ha asegurado el aneurisma puede llevar a un riesgo alto de resangrado4.
Se ha descrito un grupo de pacientes que se pueden rebeneficiar de la terapia por un tiempo más prolongado, como aquéllos con aneurismas de la arteria cerebral media, infartos cerebrales o hematomas y los que tienen historia de convulsiones previas4.
Es recomendada la utilización profiláctica de anticonvulsivos. Los medicamentos más utilizados son: fenitoína, fenobarbital y ácido valproico.
La fenitoína se inicia con un bolo de 10-15 mg/ kg, pasar a una taza de infusión no mayor de 50 mg/kg, continuar a una dosis promedio de 1,5 mg/ kg cada 8 horas por vía endovenosa. La vía oral se iniciará una vez las condiciones del paciente lo permitan. Los detractores del uso de este medicamento muestran que la droga puede estar implicada en un empeoramiento del nivel cognoscitivo de supervivientes de vasoespasmo por hemorragia subaracnoidea aneurismática83. Por esta razón, preferimos la utilización de fenobarbital, dado que este medicamento provee adecuada protección contra las convulsiones, produce grados de sedación deseables en pacientes con vasoespasmo por hemorragia subaracnoidea aneurismática, sin tener eventos adversos aparentes en el resultado neurológico de los mismos.
La Protección gástrica se instaura en todos los casos, dado los beneficios demostrados ampliamente en pacientes quirúrgicos críticos84,85.
El control de los niveles plasmáticos de glucosa son cada vez más importantes, cuando se habla del control metabólico y resultado neurológico de pacientes que cursan con cualquiera de los tipos de accidentes cerebrovasculares, incluyendo aquellos con hemorragia subaracnoidea86,87,88,89,90.
La hiperglucemia es un marcador de mortalidad y de pronóstico neurológico malo en pacientes con un ACV isquémico que no eran previamente diabéticos88. Estudios recientes muestran que la hiperglucemia en las primeras 72 horas del ingreso de los pacientes con HSA aguda, es un factor de predicción independiente de mal resultado neurológico87,89 y si bien, no es un factor de predicción demostrado de déficit isquémico tardío, si se ha encontrado que empeora el resultado neurológico en aquellos que lo desarrollan86.
La hiperglucemia en pacientes con HSA aneurismática es secundaria a una gran activación simpática. Esta activación simpática eleva las cifras glucémicas en forma directa a través de liberación de norepinefrina e indirecta por secreción de hormonas contrarreguladoras como el glucagón y la hormona del crecimiento; junto con disminución o resistencia a la insulina.
Los altos niveles de glucosa podrían ejercer efectos indeseables en pacientes con enfermedad cerebrovascular por varios mecanismos:86,87,88,89
- El aumento de la disponibilidad de glucosa en tejidos del área de penumbra isquémica, que incrementa los niveles de lactato intracelular y con ello acidosis intracelular
- Aumento en la formación de ácidos grasos libres, que favorecen la peroxidación lipídica
- Aumento en la concentración intracelular de calcio
- Pacientes con historia de disglucemia tienen disfunción endotelial y respuesta vascular anormal al estrés, en el aspecto vasomotor y en la actividad antiinflamatoria y antitrombótica.

Todos ellos nocivos para las neuronas, esto se refuerza por reportes que han mostrado disminución del infarto cerebral con la administración de insulina90.
Pero no todos los autores están de acuerdo87, algunos postulan que la hiperglucemia es simplemente una respuesta de estrés, que aumenta en la medida en que la enfermedad sea más grave. Sin embargo, existe acuerdo en que los pacientes críticos y en fase aguda sean vigilados con niveles de glucemia, al menos cada 6 horas y, luego; modificar según la evolución.
El objetivo es mantener al paciente euglucémico o con niveles de insulina menores a 120 mg/dl a través de un esquema móvil de insulina o de infusión continua si los niveles no se controlan fácilmente.
BIBLIOGRAFÍA
1. Rinkel G , Feigin V , Algra A , et al. Calcioantagonistas para la hemorragia subaracnoidea por aneurisma (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus , 2005 Número 2. Oxford: Update Software Ltd. Disponible a: www.update-software.com(Traducida de The Cochrane Library, 2005 Issue 2. Chichester , UK : John Wiley & Sons, Ltd.). [ Links ]
2. Pickard J, Murray G, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ. 1989;298:636-42. [ Links ]
3. Harrod C, Bendok B, Batjer H, et al. Prediction of cerebral vasospasm in patients presenting with aneurysmal subrachnoid hemorrhage: A review. Neurosurgery. 2005; 56:633-51 [ Links ] [ Links ]
5. Rinkel G, Feigin V, Algra A, et al. Tratamiento de expansión del volumen circulatorio para la hemorragia subaracnoidea por aneurisma (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus , 2005 Número 2. Oxford: Update Software Ltd. Disponible a: www.update-software.com (Traducida de The Cochrane Library, 2005 Issue 2. Chichester , UK : John Wiley & Sons, Ltd.). [ Links ]
6. Widjics E, Kallmes D, Manno E, et al. Subrachnoid hemorrhage: Neurointensive care and aneurysm repair. Mayo clinic proceed. 2005;80:550-9 [ Links ]
7. Treggiari M, Walder B, Suter P, et al. Systematic review of the prevention of delayed ischemic neurological deficits with hypertension, hipervolemia, and hemodilution following subarachnoid hemorrhage. J neurosurg. 2003;98:978-86. [ Links ]
8. Hoh B, Carter B, Ogilvy C. Risk of hemorrhage of unsecured, unruptured aneurysms during and after hypertensive, hypervolemic therapy. Neurosurgery. 2002;50:1207-1212. [ Links ]
9. Noro M, Katayama Y, Kojima J, et al. Prophylactic management of excessive natriuresis with hidrocortisona for efficient therapy after subarachnoid hemorrhage. Stroke. 2003;34:2807;11. [ Links ]
10. Kim D, Joseph M, Ziadi S, et al. Increases of cardiac output can reverse flow deficit from vasospasm independent of blood pressure: A study using Xenon computed tomographic measurement of cerebral blood flow. Neurosurgery. 2003;53:1044;52. [ Links ]
11. Smith T, Enterline D. Endovascular treatment of cerebral vasospasm. JVIR. 2000;11:547-58. [ Links ]
12. Muizeelar J, Zwienenberg M, Rudisill N, et al. The prophylactic use of transluminal ballon angioplasty in patients with Fisher grade 3 subarachnoid hemorrhage: a pilot study. J neurosurg.1999;91:51-8. [ Links ]
13. Claassen J, G Bernardini, Kreiter L. Effect of cisternal and ventricular blood on risk of delayed cerebral ischemia after subarachnoid hemorrhage. The Fisher scale revisited. Stroke. 2001;32:2012-20. [ Links ]
14. Dumont A, Dumont R, Chow M, et al. Cerebral vasospasm after subarachnoid hemorrhage: Putative rol of inflamation. Neurosurgery. 2003;53:123-35. [ Links ]
15. Fisher C, Kistler J, Davis J. Relation of cerebral vasospasm to subarachnoidhemorrhage visualized by computerized tomographic scanning. Neurosurgery. 1980;6:1-9. [ Links ]
16. Lisakowski C, Walder B, Costanza M, et al. Transcranial Doppler versus angiography in patients with vasospasm due to a ruptured cerebral aneurysm. A systematic review. Stroke.2001;32:2292-8. [ Links ]
17. Springdorf J, Fredericksen M, Eskessen V, et al. Trends in monitoring patients with subarachnoid hemorrhage. BJA 2005;94:259-70. [ Links ]
18. Moppet I, Mahajan R. Transcranial doppler ultasonography in anesthesia and intensive care unit. BJA 2004; 93:710-24. [ Links ]
19. Le Roux P, Elliott J, Eskridge J, et al. Risks and benefits of diagnostic angiography after aneurysm surgery: A retrospective analysis of 597 studies. Neurosurgery. 1998; 42:1248-54. [ Links ]
20. Vora Y, Suarez-Almazor M, Steinke D, et al. Role of transcranial monitoring in the diagnosis of the cerebral vasospasm after subarachnoid hemorrhage. Neurosurgery. 1999;44:1237-48. [ Links ]
21. De Gans, Niewkamp D, Rinkel G, et al. Timing of aneurysm surgery in subarachnoid hemorrhage: A systematic review of literature. Neurosurgery. 2002; 50: 336-42. [ Links ]
22. Doersch N. Therapeutic approachs to vasospasm in subarachnoid hemorrhage. Crit care med. 2002;8:128-33. [ Links ]
23. Rosenwasser R, Armonda R, Thomas J, et al. Therapeutic modalities for the management of cerebral vasospasm: timing of endovascular options. Neurosurgery 1999, 44:975-80 [ Links ]
24. Biondi A, Ricciardi G, Puybasset L, et al. Intraarterial nimodipine for symptomatic cerebral vasospasm after subarachnoid hemorrhage: Preliminary results. Am J Neurorad 2004;25:1067-76. [ Links ]
25. Oliveira-Filho J, Ezzeddine M, Segal A, et al. Fever in subarachnoid hemorrhage: Relationship to vasospasm and outcome. Neurology.2001;1299-304 [ Links ]
26. Haley E, Kassel N, Apperson-Hansen C. A randomized, double-blind, vehicle-controlled trial of tirilazad mesylate in patients with aneurysmal subarachnoid hemorrhage. A cooperative study in North America. J neurosurg. 1997; 86:467-74 [ Links ]
27. Lanzino G, Kassell N, Dorsch N, et al. Double-blind randomized, vehicle-controlled study of high-dose tirilazad mesylate in women with aneurysmal subarachnoid hemorrhage. Part 2. A cooperative study in northamerica. J neurosurg.1999;90:1011-17 [ Links ]
28. Findlay J, Kassell N, Weir K, et al. A randomized trial on intraoperative, intracisternal, tissue plasminogen activator for the prevention of vasospasm. Neurosurgery. 1995;37:168-78 [ Links ]
29. Roos Y, Rinkel G, Vermeulen M, et al. Antifibrinolytic therapy for aneurysmal subarachnoid haemorrhage. Cochrane database of systematic reviews 2000;sigue 2. Oxford: Update software. [ Links ]
30. Roos Y. Antifibrinolytic treatment in subarachnoid haemorrhage: a randomized placebo-controlled trial. STAR study group. Neurology. 2000;54:77-82 [ Links ]
31. Bambakidis N, Selman W. Subarachnoid hemorrhage en: Suarez J. Critical care neurology and neurosurgery. New Jersey. 2004; 20:365-77 [ Links ]
32. Leipzig T, Redelman K, Horner T. Reducing the risk of rebleeding befote early aneurysm surgery: a posible role for antifibrinolytic therapy. Journal neurosurg. 1997; 86:220-5 [ Links ]
33. Niskanen M, Koivisto T, Ronkainnen A, et al. Resource use after subarachnoid hemorrhage: Comparisson between endovascular and surgical treatment. Neurosurgery. 2004;54:1081-88 [ Links ]
34. Koivisto T, Nanninen N, Hurskainen H, et al. Outcomes of early endovascular versus surgical treatment of ruptured cerebral aeurysms. Stroke. 2000;31:2369-77 [ Links ]
35. Lusseveld E, Brlistra E, Nijssen P, et al. Endovascular coiling versus surgical cliping in patients with a ruptured basilar tip aneurysm. Journal of neurology, neurosurgery and psychiatry.2002;53:571-3 [ Links ]
36. Molyneux A, Kerr R, Shrimpton J, et al. International subarachnoid aneurysm trial (ISAT) of neurosurgical clipping versus endovascular coiling in 2143 patients with ruptured intracranial aneurysms: a randomized trial. Lancet.2002;360:1267-74 [ Links ]
37. Ross N, Hutchinson P, Seeley H. Timing of surgery in supratentorial aneurysmal subarachnoid haemorrhage: report for a prospective study. Journal of neurology, neurosurgery and psychiatry.2002;72:480-4 [ Links ]
38. Goddard A, Raju P, Gholkar A. Does the method of treatment of acutely ruptured intracranial aneurysms influence the incidence and duration of cerebral vasospasm and clinical outcome. J neurology, neurosurgery psychiat. 2004;75:868-72 [ Links ]
39. Paul K, Kestle J, MacDonald J, et al. Marked reduction on cerebral vasospasm with lumbar drainage of cerebrospinal fluid after subarachnoid hemorrhage. J neurosurg.2004;100;215-24 [ Links ]
40. Kasuya H, Shimizu H, Kagawa M: The effect of continuous drainage of cerebrospinal fluid in patients with subarachnoid hemorrhage: a retrospective analysis of 108 patients. Neurosurgery.1991;28:56–59 [ Links ]
41. Terada T, Kinoshita Y, Yokote H. The effect of endovascular therapy for cerebral arterial spasm, its limitations and pitfalls. Acta Neurochir 1997;139:227-34. [ Links ]
42. Hui C, Lau K. Efficacy of the intra-arterial nimodipine in the treatment of cerebral vasospasm complicating subarachnoid haemorrhage. Clin radiol.2005;60:1030-36 [ Links ]
43. Al-Yamany M, Wallace M. Management of cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage. Intensive Care Med 1999;25:1463-6. [ Links ]
44. Bracard S, Levedinsky A, Anxionnat R, et al. Endovascular treatment of Hunt and Hess grade IV and V aneurysms. AJNR.202;23:953–957. [ Links ]
45. Sloan M, Alexandrove A, Tegeler C, et al. Assesment: Transcranial doppler ultrasonography. Report of thr therapeutics and technologies assesment subcommittee of the American academy of neurology. Neurology. 2004;62:1468-81. [ Links ]
46. Mascia L, Fedorko L, Terbrugge K, et al. The accuracy of transcranial doppler to detect vasospasm in patients with aneurysmal subarachnoid hemorrhage. Intensive care medicine.2003;29:1088-94. [ Links ]
47. Vajkoczy P, Meyer B, Weidauer S, et al. Clazosentan (AXV- 034343), a selective endothelin. A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. J Neurosurg. 2005;103:9-17. [ Links ]
48. Rabinstien A, Friedman J, Nichols D, et al. Predictors of outcome after endovascular treatment of cerebral vasospasm. AJNR.2004;25:1778-82. [ Links ]
49. Reilly C, Amidey C, Tolentino J, et al. Clot volumen and clerance rate as independent predictor of vasospasm after aneurysmal subarachnoid hemorrhage. J Neurosurgery. 2004;101:255 [ Links ]
50. Mariak Z, Krejza J, Swiercz M y cols. Seguridad de la ultrasonografía doppler transcraneal para el diagnóstico de espasmo de la arteria cerebral media determinado por análisis ROC (receptor operating characteristic análisis). J Neurosurg. 2002; 96: 323–30. [ Links ]
51. Okada Y, Shima T, Nishida M, et al. Comparison of transcranial Doppler investigation of aneurysmalvasospasm with digital subtraction angiographic and clinical findings. Neurosurgery 1999; 45:443–50 [ Links ]
52. Suarez J, Qureshi A, Yaia A, et al. Symptomatic vasospasm diagnosis alter subarachnoid hemorrhage: evaluation of transcranial doppler ultrasound and cerebral angiography as related to compromised vascular distibution. Crit care Med. 2002;30:1348-55 [ Links ]
53. Newell D, Grady M, Eskridge J, et al. Distribution of angiographic vasospasm after subarachnoid hemorrhage: implications for diagnosis by transcaranial Doppler ultrasonography. Neurosurgery.1990;27:574-7 [ Links ]
54. Ekelund A, Saveland H, Romner B, et al. Is transcraneal Doppler sonogrphy useful in detecting late cerebral ischaemia after aneurysmal subarachnoid haemorrhage?. Br J Neurosurg.1996;10:19-25 [ Links ]
55. Grosset D, Straiton J, du T, et al. Prediction of symptomatic vasospasm after subarachnoid hemorrhageby paridly increasing transcranial Doppler velocity and cerebral blood flow changes. Stroke.1992;23:674-9 [ Links ]
56. Polin R, Coenen V, Hansen C, et al. Efficacy of transluminal angioplasty for the management of symptomatic cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg 2000;92:284–90 [ Links ]
57. Murayama Y, Song J, Uda K, et al. Combined endovascular treatment for both intracranial aneurysm and symptomatic vasospasm. 2003. AJNR; 24:133–139 [ Links ]
58. Badjiatia N, Topcouglu M, Pryor J, et al. Preliminary experience with intra-arterial nicardipine as a treatment for cerebral vasospasm. AJNR 2004; 25:819–826 [ Links ]
59. Feng l, Fitzimmons B, Young W, et al. Intraarterially administred verapamil as adjunct therapy for cerebral vasospasm: Safety and 2-year experience. AJNR. 2002; 23:1284–90 [ Links ]
60. Andaluz N,Tomsick B, TewJ, et al. Indications for endovascular therapy for refractory vasospasm after aneurysmal subarachnoid hemorrhage. Experience at the Cincinatti University. Surg Neurol. 2002;58:131–8 [ Links ]
61. Solomon R, Fink M, Lennihan L. Early aneurysm surgery and prophylactic hypervolemic hypertensive therapy for the treatment of aneurysmal subarachnoid hemorrhage. Neurosurgery. 1988;23:699–70 [ Links ]
62. Eskridge J, McAuliffe, Song J, et al. Balloon angioplasty for the treatment of vasospasm: Results of first 50 cases. Neurosurgery 1998;42:510–7 [ Links ]
63. Johnston S. Effect of endovascular services and hospital volume on cerebral aneurysm treatment outcomes. Stroke 2000;31:111–17 [ Links ]
64. Polin R, Apperson H, German P, et al. Intra-arterially administered papaverine for the treatment of symptomatic cerebral vasospasm. Neurosurgery. 1998;42:1256–67 [ Links ]
65. Elliott J, Newell D, Lam D, et al. Comparison of balloon angioplasty and papaverine infusion for the treatment of vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg 1998;88:277–84 [ Links ]
66. Schuknecht B, Fandino J, Yüksel C, et al. Endovascular treatment of cerebral vasospasm: assessment of treatment effect by cerebral angiography and transcranial colour Doppler sonography. Neuroradiology.1999;41: 453-62 [ Links ]
67. Kassel N Torner J, Haley E, et al. The international cooperative study on the timing of aneurysm surgery. J Neurosurg.1990;73:18–36 [ Links ]
68. Turowski B, du Mesnil de Rochemont R, Beck J, et al. Assesment of changes in cerebral circulation time due to vasospasm in a specific arterial territory: effect of angioplasty. Neuroradiology.2005;47: 134–143 [ Links ]
69. Prat R, Misra M, Dujovny M. Endovascular treatment of vasospasm following subarachnoid hemorrhage. Crit Rev Neurosurg. 1997;7:149–55 [ Links ]
70. Zubkov Y, Alexander l, Smith, et al. Angioplasty of vasospasm: is it reasonable? Neurol Res. 1994;16:9-11. [ Links ]
71. Oskouian R, Martin N, Hong J, et al. Multimodal quantitation of the effects of endovascular therapy for vasospasm on cerebral blood flow, transcranial doppler ultrasonographic velocities, and cerebral artery diameters. Neurosurgery 2002;51:30-43 [ Links ]
72. Reill C, Amidei C, Tolentino J, et al. Clot volume and clearance rate as independent predictors of vasospasm after aneurysmal subarachnoid hemorrhage. J Neurosurg. 2004;101:255–261 [ Links ]
73. Mayberg H, Batjer H, Dacey R, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage. A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Circulation.1994;90:2592-605 [ Links ]
74. Kassell N, Torner J, Haley E, et al. The International Cooperative Study on the Timing of Aneurysm Surgery I: overall management results. J Neurosurg. 1990;73:18- 36. [ Links ]
75. Kassell N, Torner J, Jane J, et al. The International Cooperative Study on the Timing of Aneurysm Surgery II: surgical results. J Neurosurg. 1990;73:37-47 [ Links ]
76. Darlong V, Jayalakhsmi T, Kaul H. Stress ulcer prophylaxis in patients on ventilator. Trop Gastroenterol. 2003;24(3):124-8 [ Links ]
77. Cook D, Guyatt G, Marshall J, et al. A comparison of sucralfate and ranitidine for the prevention of upper gastrointestinal bleeding in patients requiring mechanical ventilation. Canadian Critical Care Trials Group. N Engl J Med.1998;338:791-7 [ Links ]
78. Hoh B, Ogily C. Endovascular treatment of cerebral vasospasm: Transluminal balloon angioplasty. intraarterial papaverine and intra-arterial nicardipine. Neurosug Clin N Am. 2005;16:501-516 [ Links ]
79. Zubkov A, Lewis A, Scalzo D, et al. Morphologic changes after percutaneus transluminal angioplasty. Surg Neurol. 1999;51:399-403 [ Links ]
80. Mejía J, Pernow J, Van Holst H, et al. Effects of neuropeptide Y, calcitonina gene-related peptide, substance P and capsaicin on cerebral arteries in man and animals. J Neurosurgery 1988;69:913-18 [ Links ]
81. McGirt M, Mavrapoulus J, McGirt L, et al. Leukocytosis as an independent risk factor for cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg. 2003;98;1222-26 [ Links ]
82. Dumont A, Dumont R, Chow M, et al. Cerebral vasospasm alter subarachnoid hemorrhage: Putative rol of inflammation. Neurosurgery. 2003;53:123-35 [ Links ]
83. Lobato R, Marin J, Salaices M, et al. Effect of experimental subarachnoid hemorrhage on the adrenergic inervation of cerebral arteries. J Neurosurg. 1980;53: 477-9 [ Links ]
84. Lobato R, Marin J, Salaices M, et al. Cerebrovascular reactivity to adrenaline and serotonin following experimental subarachnoid hemorrhage. J Neurosurg. 1980; 53:480-5 [ Links ]
85. Delgado T. Central Cathecolamine pathways involved in cerebral vasospasm and changes in cerebral blood flow and glucose metabolism after a subarachnoid haemorrhage. An experimental study in the rat. University of Lund. 1986;1:9-15 [ Links ]
86. Voldby B, Enelvodsen E, Jensen F. Regional CBF, intraventricular pressure, and cerebral metabolism in patients with ruptured intracraneal aneurysms. J Neurosug. 1985;62:48-58 [ Links ]
87. Svendgaaard N, Brismar J, Deldago T, et al. Effect on the development of vasospasm of selective lesions of the catecholamine systems in the brain stem. Stroke. 1985;16:602-8 [ Links ]
88. Fassbender K, Hodapp B, Rossol S, et al. Inflammatory cytokines in subarachnoid hemorrhage: Association with abnormal blood flow velocities in basal cerebral arteries. J Neurol Neurosurg Psych 2001;70:534–7 [ Links ]
89. Mori T, Katayama Y, Kawamata T, et al. Improved efficiency of hypervolemic therapy with inhibition of natriuresis by fludrocortisone in patients with aneurismal subarachnoid hemorrhage. J Neurosurg 1999;91:947-52 [ Links ]
90. Kassell N, Peerless S, Durward Q, et al. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982;11:337-43 [ Links ]
91. Naidech A, Kreiter K, Janjua N, et al. Phenytoin exposure is associated with functional and cognitive disability after subarachnoid hemorrhage. Stroke 2005; 36:583-7 [ Links ]
92. Juvela S, Siironen J, Huhmonen J. Hyperglycemia, excess weight, and history of hipertensión as risk factors for poor outcome and cerebral infaction alter aneurysmal subarachnoid hemorrhage. J Neurosurg 2005;12:1003 [ Links ]
93. Claassen J, Vu A, Kreiter K, et al. Effect of acute physiologic derangements on outcome alter subarachnoid hemorrhage. Crit Care Med 2004; 32:832-8 [ Links ]
94. Capes S, Hunt D, Malmberg K, et al. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients. Stroke 2001;32:2426-32 [ Links ]
95. Dorhout S, van Dijk G, Algra A, et al. Glucosa levels and outcome after subarachnoid hemorrhage. Stroke 2003; 61:1132-3 [ Links ]
96. Van den Berghe G, Wouters P, Weekers F, et al. Intensive insulin therapy in the critically ill patients. N Engl J Med 2001;345:1359-67. [ Links ]
ALGORITMO DE TRATAMIENTO
HEMORRAGIA SUBARACNOIDEA ANEURISMÁTICA (HSA)
1. Aneurisma no clipado, fase aguda de HSA:
Monitoría Invasiva con catéter venoso central, línea arterial y catéter vesical
A) Tratamiento farmacológico:
1. Mantener normovolemia. PVC entre 8-10mmHg
2. Mantener normonatremia con medición diaria de sodio sérico
3. Mantener presión arterial media (PAM) mayor de 90 a 100 mmHg
4. Minimizar el uso de hipotensores o sedantes
5. Nimodipino oral 60 mg VO cada 4 horas x 21 días o endovenoso de 0,5 a 2 mg/h, estricto control de tensiones arteriales
6. Protección gástrica con ranitidina 50 mg IV cada 8 horas
7. En caso de hipotensión aplique al algoritmo de hipotensión
8. Fenobarbital 100 mg iv cada 8 horas o 50 mg vo cada 12 horas o Fenitoína 15 a 20 mg/k iv diluidos en solución salina normal a una taza de infusión no mayor a 50 mg/min y cointinuar 100 mg cada 8 horasª.
B) Apoyo diagnóstico:
1. Obtener tomografía computada (TC) de cerebro simple
2. Doppler transcraneal desde el inicio y cada segundo día
3. Arteriografía cerebral
2. Aneurisma no clipado, ingreso tardío sin vasoespasmo
A) Tratamiento farmacológico:
1. Mantener normovolemia. PVC entre 8- 10mmHg
2. Mantener normonatremia, medición de sodio sérico cada 12 horas
3. Mantener presión arterial media (PAM) mayor de 100 mmHg
4. Evitar hipotensores o sedantes
5. Nimodipino oral 60 mg VO cada 4 horas por 21 días o endovenoso de 0,5 a 2 mg/h, estricto control de tensiones arteriales
6. En caso de hipotensión aplique el algoritmo de hipotensión
7. Protección gástrica con ranitidina 50 mg cada 8 horas IV
8. Fenobarbital 100mg iv cada 8 horas o fenitoína 15 a 20 mg/k iv diluidos en solución salina normal a una taza de infusión no mayor a 50 mg/min y cointinuar 100 mg cada 8 horas*
B) Apoyo diagnóstico:
1. Obtener tomografía computada (TC) de cerebro simple
2. Doppler transcraneal desde el inicio y cada segundo día
3. Arteriografía cerebral
3. Aneurisma no clipado, ingreso tardío con vasoespasmo clínico
A) Tratamiento farmacológico:
1. Mantener normovolemia. PVC de 10 y POAP de 12 a 14 mm Hg
2. Mantener normonatremia, medición de sodio sérico cada 12 horas
3. Mantener presión arterial media (PAM) mayor de 100 mmHg
4. Evitar hipotensores o sedantes
5. Nimodipino oral 60 mg VO cada 4 horas por 21 días o endovenoso de 0,5 a 2 mg/h, estricto control de tensiones arteriales
6. En caso de hipotensión aplique al algoritmo de hipotensión
7. Protección gástrica con ranitidina 50 mg cada 8 horas IV
8. Fenobarbital 100 mg iv cada 8 horas o 50 mg vo cada 12 horas o Fenitoína 15 a 20 mg/k iv diluidos en solución salina normal a una taza de infusión no mayor a 50 mg/min y cointinuar 100 mg cada 8 horas.
B) Apoyo diagnóstico:
1. Obtener tomografía computada (TC) de cerebro simple
2. Doppler transcraneal desde el inicio y cada segundo día
3. Arteriografía cerebral
C) Intervención quirúrgica y/o endovascular
1. Considerar tratamiento endovascular para vasoespasmo y para el aneurisma al mismo tiempo
2. Considerar tratamiento endovascular para vasoespasmo inicial y tratamiento tardío para el aneurisma por clipaje o por terapia endovascular.
ALGORITMO DE INTERVENCIÓN PARA HIPOTENSIÓN
En pacientes hipotensos con TAS menor de 90 mmHg al ingreso iniciar:
1. Líquidos intravasculares como cristaloides o coloides a una dosis de 3 a 7 ml/kg según el estado clínico y repetir hasta PVC de 8 a 10 mmHg
2. Si no se consiguen las metas propuestas en el algoritmo de intervención de HSA-vasoespasmo iniciar dopamina 5 a 10 mcg/kg/m
3. Si no se logran las metas propuestas en el algoritmo de intervención de HSA-vasoespasmo iniciar noradrenalina 0,05 a 0,3 mcg/kg/m o fenilefrina 10 a 30 mcg/k/h.
4. Aneurisma clipado
Clasificación según estratificación de riesgo (escalas de Fisher):
1. Pacientes de bajo riesgo:
1.1 Mantener PVC entre 8-10 mmHg
1.2 Mantener PAM entre 100-120 mmHg
1.3 Nimodipino 60 mg VO cada 4 horas por 21 días o endovenoso de 0,5 a 2 mg/h, estricto control de tensiones arteriales
1.4 Hemodilución hasta hematocrito de 30-40% mínimo en tres días
1.5 Protección gástrica con ranitidina 50 mg cada 8 horas IV
1.6 Fenobarbital 100 mg iv cada 8 horas o 50 mg vo cada 12 horas o Fenitoína 15 a 20 mg/k iv diluidos en solución salina normal a una taza de infusión no mayor a 50 mg/ min y cointinuar 100 mg cada 8 horas, monitoreo de niveles plasmáticos cuando el clínico lo considere adecuado.
1.7 Doppler transcraneano cada segundo día
2. Pacientes de alto riesgo:
2.1 Insertar catéter en arteria pulmonar.
2.2 Obtener presión de oclusión en la arteria pulmonar (POAP) entre 14 y 16 mm Hg
2.3 Tensión arterial sistólica (TAS) entre 150- 170 mmHg, si se necesita iniciar dopamina < 10 mcg/kg/minuto
2.4 Nimodipino 60 mg VO cada 4 horas por 21 días endovenoso de 0,5 a 2 mg/h, estricto control de tensiones arteriales
2.5 Hemodilución hasta hematocrito de 30-40% mínimo en tres días
2.6 Protección gástrica con ranitidina 50 mg cada 8 horas IV
2.7 Fenobarbital 100 mg iv cada 12 horas o 50 mg vo cada 12 horas o Fenitoína 15 a 20 mg/k iv diluidos en solución salina normal a una taza de infusión no mayor a 50 mg/ min y cointinuar 100 mg cada 8 horas, monitoreo de niveles plasmáticos cuando el clínico lo considere adecuado.
2.8 Doppler transcraneano cada día o cada segundo día según disponibilidad
3. Pacientes que desarrollan vasoespasmo clínico
3.1 Optimizar POAP idealmente de 16 a 18 mm Hg con el volumen necesario de líquidos endovenosos (coloides o cristaloides), si se encuentra diuresis mayor a 250 ml /hora iniciar vasopresina 3 a 5 ui cada 8 a 6 horas sc dea cuerdo a necesidad ofludrocortisona 0.1 -0.2 mg VO cada 12 horas o hidrocortisona 100 mg IV cada 8 horas, en ese orden.
3.2 Evaluar estado clínico en 1 hora
3.3 Si no hay mejoría clínica llevar TAS > 180 mm Hg con vasopresores, iniciar noradrenalina 0.05-0.2mcg/kg/minuto o fenilefrina con control cercano del estado clínico
3.4. Evaluar estado clínico en 1 hora *
3.5 Si no hay mejoría clínica iniciar inodilatadores como dobutamina 0.2 - 0.5 mc/kg/ minuto o milrinone 0.375 - 0.75 mcg/kg/ minuto hasta lograr un índice cardíaco mayor a 3.5 L/minuto/m2
3.6 Evaluar estado clínico en 1 hora
3.7 Si no hay mejoría clínica, continuarán las medidas instaladas pero además se llevará a terapia endovascular lo más temprano posible (primeras 24 horas) con angioplastia mecánica o química con nimodipina intraarterial según tome la decisión el servicio de neuroradiología.
3.8 Se tomará doppler transcraneal de seguimiento cada día o segundo día según disponibilidad y TC o angiografía de acuerdo a la condición clínica del paciente
3.9 Se completará el tratamiento con nimodipina 60 mg VO cada 4 horas hasta el día 21 del sangrado inicial o endovenoso de 0,5 a 2 mg/h, estricto control de tensiones arteriales
3.10 Protección gástrica con ranitidina 50 mg IV cada 8 horas
3.11 Fenobarbital 100 mg iv cada 8 horas o 50 mg vo cada 12 horas o Fenitoína 15 a 20 mg/k iv diluidos en solución salina normal a una taza de infusión no mayor a 50 mg/min y cointinuar 100 mg cada 8 horas, monitoreo de niveles plasmáticos cuando el clínico lo considere adecuado.
