










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombian Journal of Anestesiology
Print version ISSN 0120-3347
Rev. colomb. anestesiol. vol.36 no.3 Bogotá July/Sep. 2008
ARTÍCULO DE REVISIÓN
Trastornos de coagulación en trauma craneoencefálico
M. C. Niño de Mejía1, M. V. Caicedo2, J. A. Torres3, J. A. Tovar4.
1. Neuroanestesióloga, Hospital Universitario Fundación Santa Fe de Bogotá, Bogotá, D.C., Colombia Email:gigi87@yahoo.com
2. Residente de anestesia, Hospital Universitario Fundación Santa Fe de Bogotá, Bogotá, D.C., Colombia
3. Residente de anestesia, Hospital Simón Bolívar, Universidad El Bosque, Bogotá, D.C., Colombia
4. Residente de anestesia, Universidad Surcolombiana, Neiva, Colombia
Recibido para publicación agosto 21 de 2008, Aceptado para publicación septiembre 22 de 2008
RESUMEN
La comprensión del proceso de la coagulación ha progresado durante la última década, evolucionando a partir del concepto según el cual la producción del coágulo se iniciaba por acción de las plaquetas y la activación de uno de los dos sistemas separados, la vía extrínseca y la vía intrínseca, al concepto actual que hace énfasis sobre la vía común y un sistema proteolítico que da lugar a la degradación de los coágulos formados y a la prevención de la formación indeseada de coágulos.
La alteración de este equilibrio cobra especial importancia en los pacientes con trauma craneoencefálico, en quienes -a la luz de los conocimientos actuales- se pueden presentar trastornos de la coagulación que van desde lesiones procoagulantes, en un extremo, hasta lesiones anticoagulantes, en el otro extremo. La meta de los autores es brindar a los clínicos de una guía de evaluación inicial, de seguimiento y, de las posibilidades terapéuticas disponibles en el momento.
Palabras clave: Trauma craneoencefálico, trastornos procoagulantes y anticoagulantes, evaluación inicial.
ABSTRACT
The understanding of the coagulation process has progressed during last decade evolving from the concept according to which the production of the clot begins by means of the action of platelets and the activation of one of two separated systems, the extrinsic route and the intrinsic route, to the present concept that makes emphasis on the common route and a proteolytic system that give rise to the degradation of formed clots and to the undesired prevention of the formation of the clot. The alteration of this balance receives special importance in the patients with brain trauma in those who to the light of the present knowledge may present upheavals of the coagulation which can go from procoagulating injuries in one end to anticoagulating injuries in the other. The goal of the authors is to provide a clinical guide with initial evaluation, pursuit and therapeutic possibiliti.es available at the moment.
Key words: Craneoencephatic trauma, procoagulant and anticoagulant injuries, clinical initial evaluation.
INTRODUCCIÓN
Es frecuente que, tras una reanimación agresiva con líquidos endovenosos y hemoderivados en pases frecuente que, tras una reanimación agresiva con líquidos endovenosos y hemoderivados en pacientes que han sufrido un trauma grave, se produzca un sangrado persistente en áreas lesionadas y no lesionadas, el cual conlleva inevitablemente a la muerte si no es posible controlarlo. Esto se conoce como la coagulopatía del trauma.
La respuesta hemostática normal requiere la presencia y el funcionamiento adecuados de tres componentes indispensables: la pared del vaso sanguíneo, las proteínas plasmáticas y las plaquetas1. En primer lugar, está la pared del vaso sanguíneo, la cual está compuesta por células endoteliales; debajo de ellas encontramos una matriz proteica subendotelial de fibras de colágeno y fibronectina y células mesoteliales (fbroblastos y células musculares lisas). Estas células mesoteliales tienen la capacidad de expresar en grandes cantidades el factor tisular, que es el mayor iniciador de la coagulación plasmática.
En condiciones normales, las células endoteliales mantienen el equilibrio del flujo sanguíneo mediante la secreción de varias sustancias: los factores anticoagulantes como el óxido nítrico, que previene la activación plaquetaria; el activador del plasminógeno tisular, cuya función es romper los enlaces de fibrina que se hayan formado; el sulfato de heparina, que sirve como cofactor de la antitrombina III, y la trombomodulina, para la activación de la proteína C1.
Cuando las células endoteliales son activadas se revierten todos estos mecanismos y sintetizan factores procoagulantes, como el inhibidor del activador del plasminógeno. Ante un daño tisular, el colágeno expuesto con cargas eléctricas negativas se une al factor de von Willebrand y, entonces, las plaquetas pueden adherirse y pasar a su estado activo1.
El segundo componente de la respuesta hemostática son las proteínas plasmáticas, que incluyen factores de coagulación, proteínas anticoagulantes y el sistema fibrinolítico. Brevemente recordemos el antiguo modelo de coagulación (figura 1), concebido in vitro en 1964 por Davie y Ratnoff2. En él interviene la cascada de activación de los factores de coagulación, en dos posibles vías: la vía intrínseca y la vía extrínseca, las cuales finalmente convergen en una vía común para la producción de trombina (factor IIa), indispensable para la transformación del fibrinógeno en fibrina.
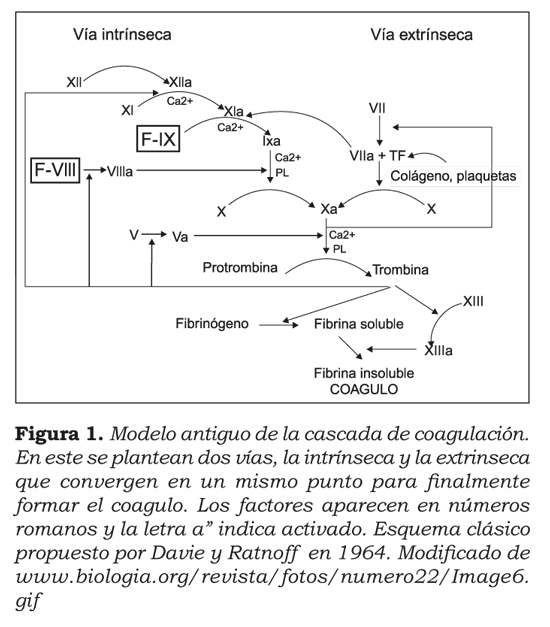
En la actualidad, se ha propuesto que la iniciación de la cascada de coagulación viene siendo determinada por la exposición del factor tisular y su unión con el factor VIIa3, que forman un complejo que activa los factores X y IX. El factor Xa activa la protrombina y produce trombina. El complejo factor tisular-VIIa-Xa, rápidamente es inactivado por el inhibidor de la vía del factor tisular; la pequeña cantidad de fibrina producida es suficiente para activar los trombocitos1 (figura 2).
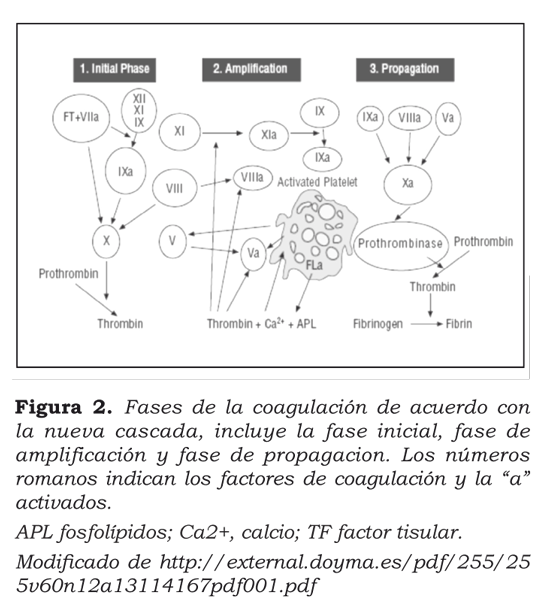
El tercero y último componente, las plaquetas, son inhibidas o activadas por los mecanismos ya referidos, con la capacidad intrínseca de producir algunos factores de coagulación.
FISIOPATOLOGÍA DE LAS ALTERACIONES DE LA COAGULACIÓN EN TRAUMA
El sangrado anormal es reconocido clínicamente cuando las mucosas, las superfcies serosas y los sitios de punción sangran por periodos prolongados, así como por la aparición de hematomas en sitios no lesionados. Teniendo en cuenta que no siempre están disponibles los estudios de laboratorio, en caso de urgencia debemos mantenernos alertas, haciendo uso racional de los datos clínicos que aportan los pacientes.
Lógicamente, debemos tener en cuenta aquellos trastornos previos de la coagulación, datos que nos provee una adecuada historia clínica, excluyendo de esta manera trastornos congénitos o adquiridos de la coagulación, así como los producidos de forma iatrogénica por medicamentos.
Varios autores han identificado la tríada de la muerte, compuesta por hipotermia, acidosis y coagulopatía1,4, la que sin duda alguna juega un papel importante en la supervivencia de los pacientes críticos.
En casos de trauma surge el concepto de circulo vicioso del sangrado4, que hace referencia a la hemorragia profusa que presentan los pacientes al sufrir un impacto, quienes posteriormente se someten a reanimación agresiva con líquidos o productos sanguíneos y alcanzan niveles de hemodilución deletéreos con el progresivo desarrollo de coagulopatía y sangrado continuo.
Cosgriff et al.4 realizaron una investigación durante dos años, en un centro de trauma mayor (Denver Health Medical Center), en pacientes que recibían más de 10 unidades de glóbulos rojos, y observaron algunos factores de riesgo involucrados en la coagulopatía del trauma.
De estos factores, los más importantes que arroja el estudio son el valor del pH sanguíneo, la temperatura corporal central, la gravedad de la lesión y la presencia de hipotensión arterial.
El valor del pH sanguíneo es el factor pronóstico más importante de coagulopatía. Meng et al.5 reportaron una disminución en la actividad del factor VIIa, del complejo VIIa/factor tisular y del complejo Xa/Va, cuando el pH se aproximaba a valores de 7. La actividad enzimática de los factores de coagulación se encuentra disminuida hasta en 90% a este pH.
El aumento en las cargas de hidrogeniones interfiere con las interacciones iónicas de los factores de coagulación, así como su interacción con los fosfolípidos de membrana plaquetarios1.
La temperatura es el segundo factor más importante. Las enzimas implicadas en la coagulación se ven afectadas, cerca de 10% por cada grado centígrado que disminuye la temperatura. Las plaquetas son las células más sensibles a los cambios de temperatura, reduciendo notoriamente su actividad. La interacción del factor de von Willebrand con las glicoproteínas Ib/IX depende en gran medida de la temperatura6.
La gravedad de la lesión es el tercer hallazgo más importante en el estudio de Cosgriff. El traumatismo del tejido cerebral, las fracturas óseas y la presencia de líquido amniótico, son situaciones clínicas en las que existe la posibilidad de embolismo, con los agravantes de que estos materiales son fuentes de tromboplastinas, contienen factor tisular y, pueden causar coagulación intravascular diseminada, consumiendo los factores de coagulación al interactuar con los mismos; agravado por el agotamiento de la antitrombina III en un intento infructuoso de contrarrestar el proceso, con la consiguiente formación de trombo y émbolos secundarios que interrumpen el flujo sanguíneo y llevan a isquemia de los tejidos. Estos procesos están directamente relacionados con la magnitud de la lesión.
Por último, tenemos el cuarto factor ligado a la coagulopatía, la hipotensión arterial, la cual representa básicamente hipovolemia, que signifca pérdida sanguínea continua a pesar de la transfusión masiva.
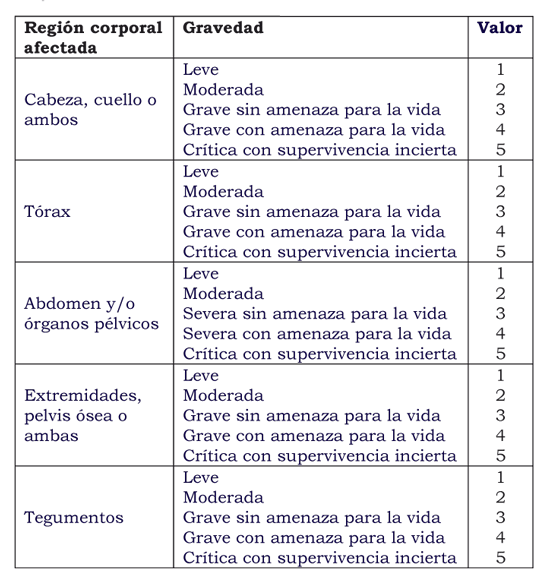
Estos factores de riesgo fueron tabulados y se encontró que el riesgo de desarrollo de coagulopatía era más alto si el pH disminuía por debajo de 7,1, la temperatura central bajaba a menos de 34ºC, la presión arterial sistólica era menor de 70 mm Hg, y la gravedad de la lesión estaba por encima de 25. En este estudio publicado por Baker et al7, se reporta que los pacientes sin ningún factor de riesgo tenían 1% de probabilidades de presentar coagulopatía; aquellos con un factor, tenían 10% a 40% de posibilidad de coagulopatía, y aquéllos con 4 factores tenían 98% de riesgo de desarrollar coagulopatía (ver cuadro).
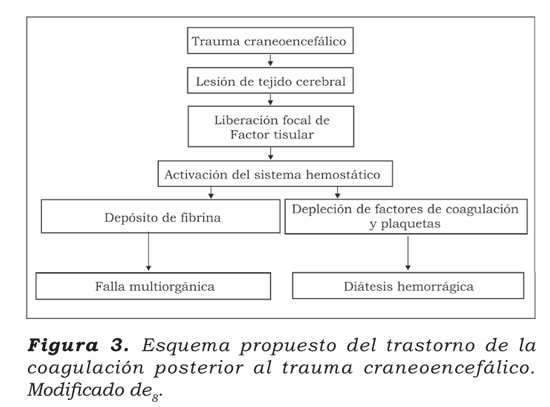
FISIOPATOLOGÍA DE LAS ALTERACIONES DE LA COAGULACIÓN EN TRAUMA CRANEOENCEFÁLICO
En individuos sanos, el equilibrio entre coagulación y fibrinólisis previene la hemorragia excesiva o la trombosis8. Los pacientes con trauma craneoencefálico están en riesgo de desarrollar alteraciones en la coagulación y en la fibrinólisis9,10,12. La causa de las alteraciones en la coagulación difiere en los pacientes con trauma craneoencefálico aislado cuando se compara con los pacientes con politraumatismo (Figura 3).
El estudio publicado por Scherer12 muestra la relación entre la extensión del tejido cerebral lesionado y la incidencia de las alteraciones de la coagulación, la cual juega un papel más importante, inclusive, que la hipoxia y el shock.
El factor tisular, principal iniciador fisiológico de la coagulación11, es liberado de forma masiva por el tejido cerebral lesionado, fenómeno que se ha postulado como la principal causa del trastorno de coagulación14-16. Gando17 muestra en su trabajo niveles de factor tisular más altos en pacientes con trauma craneoencefálico que en pacientes con otro tipo de trauma. Además, Pathak18 demostró un aumento en la actividad del factor tisular en pacientes con trauma craneoencefálico, en comparación con controles, lo cual resalta el papel de dicho factor en las alteraciones de la coagulación que siguen al trauma craneoencefálico.
Una exposición temprana del factor tisular al factor VII y al VIIa resulta en el complejo factor VIIa-factor tisular. Este complejo forma y genera pequeñas cantidades de factores Xa y IXa. El factor Xa, en colaboración con la superfcie de la membrana, convierte una pequeña cantidad de protrombina a trombina. La generación de estas pequeñas cantidades de trombina va a activar las plaquetas, el factor V y el VIII, los cuales proveen una superfcie óptima para que el complejo de protrombina pueda ensamblarse y generar más trombina, necesaria para la conversión de fibrinógeno a fibrina13,19. Esta activación de la coagulación que depende del factor tisular, produce trombos de fibrina microvasculares y macrovasculares.
Los mecanismos de control, incluso el inhibidor de la vía del factor tisular, el sistema de proteína C, antitrombina y glucosaminoglucanos, generalmente son insuficientes después de una exposición extensa del factor tisular8.
La coagulación intravascular diseminada, desencadenada por la activación del factor tisular, inhibe estos mecanismos antitrombóticos a través de la liberación de citocinas y up-regulation, y causan una alteración en las vías fisiológicas de la anticoagulación8. Esto puede causar necrosis y hemorragia en varios órganos y llevar eventualmente a falla orgánica múltiple20-23.
DISFUNCIÓN PLAQUETARIA EN TRAUMA CRANEOENCEFÁLICO
Como ya se ha descrito, la coagulopatía es un hallazgo común en el trauma craneoencefálico, se comporta como un factor de mal pronóstico y es peor si se asocia a trombocitopenia24,25. Aún no es claro si el trauma craneoencefálico y el politrauma comparten la misma fisiopatología, puesto que factores como la acidosis, la hipotermia y la hemodilución, muy frecuentes en el trauma general, rara vez se asocian con el trauma craneoencefálico26,27.
Jacoby examina 100 pacientes politraumatizados, con trauma craneoencefálico y sin él; en el subgrupo con trauma craneoencefálico, encuentra una alteración de la función plaquetaria que se asocia con un incremento en la mortalidad y se comporta como un factor de pronóstico negativo28,29,30, aun cuando el recuento plaquetario es normal.
Nekludov24 toma cuatro grupos de pacientes, en los cuales estudia el comportamiento de las plaquetas a través de la realización de tromboelastografía y mapeo plaquetario: 20 pacientes con trauma craneoencefálico grave, con escala de coma de Glasgow menor de 8; 10 pacientes politraumatizados sin trauma craneoencefálico; 7 pacientes con antecedentes de alcoholismo justificado por el hecho de que muchos pacientes con trauma craneoencefálico tenían antecedente de consumo crónico o bajo el efecto de alcohol31, y 10 voluntarios sanos. En este estudio se excluyen pacientes con antecedentes de alteraciones de la coagulación o uso de anticoagulantes; midiendo la hemoglobina, el hematocrito, el tiempo de sangría por el método de Ivy32,33, tiempos de coagulación, dímero D, fibrinógeno y marcador de daño cerebral Sb100; complementado con tromboelastografía con mapeo plaquetario.
La tromboelastografía convencional permite el estudio de las propiedades viscoelásticas de la sangre y se realiza in vitro, empleando una copa en la cual se depositan 0,36 ml de sangre. La muestra se somete a un proceso de rotación y de oscilación, evaluando las distintas etapas de la coagulación, es decir, la formación de la fibrina, la retracción del coágulo, la agregación plaquetaria y la lisis del coágulo34. El mapeo plaquetario permite la medición de la activación plaquetaria por adenosín difosfato (ADP) y ácido araquidónico; lo que permite diagnosticar de forma precisa alteraciones en la función plaquetaria (Figura 4).
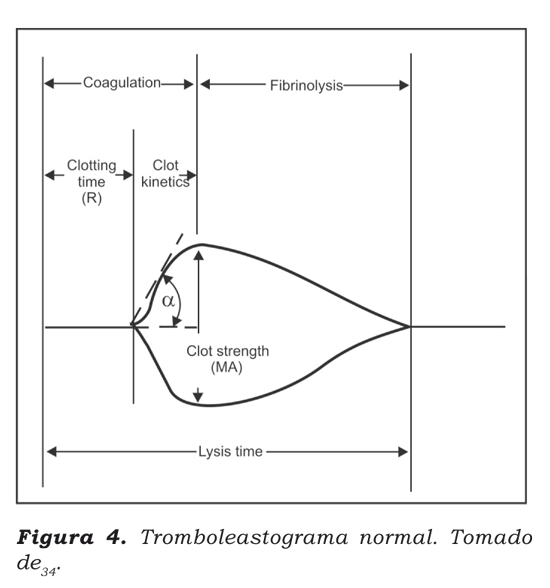
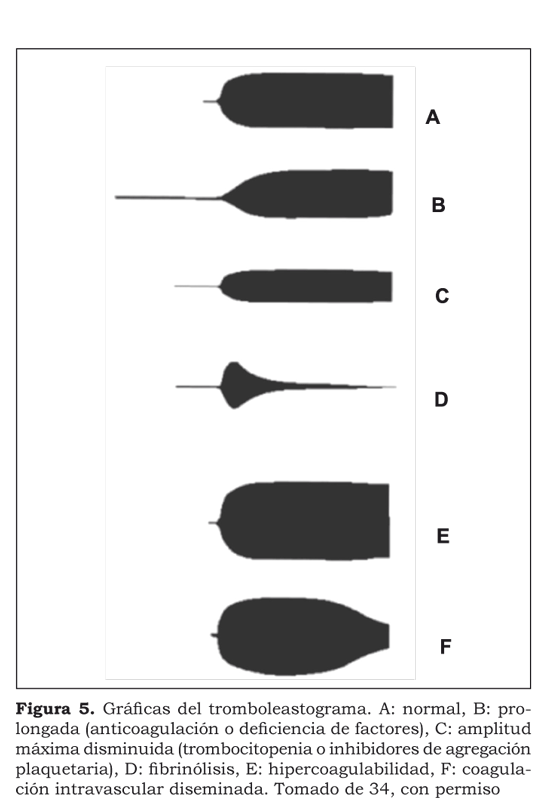
Se procesaron cuatro muestras. En la primera se realizó una tromboelastografía convencional, midiendo la MA-trombina que refeja las plaquetas activadas por la trombina. A cada una de las tres muestras restantes se les agregó heparina para inhibir la acción de la trombina. A la segunda muestra se le agregó reptilasa, enzima análoga a la trombina aislada del veneno de la serpiente Bothrops atrox y que no tiene actividad sobre las plaquetas; se diferencia de la trombina porque no es sensible a la heparina y es capaz de producir interacción trombina-fibrinógeno, aun cuando la misma trombina es neutralizada por la heparina. Esta enzima convierte el fibrinógeno en fibrina sin activar las plaquetas y muestra la MA-fibrina que mide sólo la actividad de la fibrina. A la tercera muestra se le agregó ADP, obteniendo la MAADP que evalúa la respuesta plaquetaria al ADP. A la cuarta muestra se le adicionó ácido araquidónico; el resultado de la MA-ácido araquidónico muestra la respuesta plaquetaria al ácido araquidónico24.(Figura 5).
Los resultados muestran un aumento en el tiempo de sangría y una disminución en la respuesta plaquetaria al ácido araquidónico en el grupo del trauma craneoencefálico; además, en este mismo grupo, se evidencia una disminución mayor de la respuesta plaquetaria al ácido araquidónico en los pacientes con trauma craneoencefálico que desarrollaron complicaciones hemorrágicas, con una diferencia estadísticamente significativa, lo cual muestra una alteración de los receptores plaquetarios a la cicloxigenasa o al tromboxano A2 en los pacientes con trauma craneoencefálico. La disminución en la respuesta plaquetaria al ADP, tanto en los pacientes con trauma craneoencefálico como en los pacientes politraumatizados, no es estadísticamente significativa, ni equiparable a la diferencia mostrada con el ácido araquidónico. Ambas respuestas se normalizaron a los tres días, en promedio.
Aunque se necesitan más estudios, con un mayor número de pacientes y mejor diseño metodológico, los autores plantean algunas teorías. La primera es que el trauma craneoencefálico produce la liberación de una sustancia que alteraría la función de las plaquetas; otra es que se presente una importante activación plaquetaria al momento del paso de éstas por el sitio del trauma con agotamiento de los mediadores y se desarrolle un estado refractario posterior35,36; y, finalmente, que se podría predecir que pacientes estan en riesgo de presentar complicaciones hemorrágicas posteriores.
INCIDENCIA DE LAS ALTERACIONES DE LA COAGULACIÓN
La incidencia de coagulopatía en el trauma craneoencefálico es alta, pero los valores varían según los estudios. Esta diferencia se debe a la variación en las definiciones y técnicas para el diagnóstico de coagulación intravascular diseminada. En resumen, uno de cada 3 pacientes con trauma craneoencefálico tiene signos de coagulopatía8, y ésta aumenta hasta casi 60% en trauma craneoencefálico grave37-39. En trauma craneoencefálico leve, probablemente la coagulopatía se presenta en menos de 1%40.
La coagulación intravascular diseminada parece ocurrir de manera más frecuente en pacientes con hematoma subdural agudo o contusiones parenquimatosas41.
GRAVEDAD DEL TRAUMA CRANEOENCEFÁLICO Y COAGULOPATÍA
Los trastornos de hipercoagulabilidad posteriores a trauma craneoencefálico son más prevalentes durante las primeras 24 horas y más graves en mujeres42.
Los niños con puntaje de Glasgow menor de 14, tienen un alto riesgo de presentar coagulopatía, el cual aumenta con puntajes menores43.
Stern et al.44 encontraron que casi 50% de los pacientes con trauma craneoencefálico que desarrollaban lesiones cerebrales tardías presentaban alteraciones de la coagulación, en comparación con aquéllos sin este tipo de lesión (55% Vs. 9%).
PRONÓSTICO
La presencia de coagulopatía no sólo se relaciona con el riesgo de desarrollar una lesión tardía, sino que también está relacionada con un pobre desenlace8.
El sangrado continuo, la coagulación intravascular diseminada y la isquemia cerebral secundaria a ésta, son responsables de un mal pronóstico.
El riesgo de mortalidad en pacientes con coagulopatia después de trauma craneoencefálico, es aproximadamente diez veces mayor que en los que no presenten ese trastorno de la coagulación. De la misma manera, el riesgo de un mal desenlace en pacientes con coagulopatia después de trauma craneoencefálico, es más de treinta veces mayor que en aquéllos sin coagulopatía45,46.
Diferentes estudios realizados han mostrado una relación directa entre las alteraciones en pruebas de laboratorio, como valor pronóstico. Kushimute et al.47 estudiaron los productos de degradación del fibrinógeno y los productos de degradación de la fibrina. Olson et al.45 y Selladura et al.46 encontraron que los niveles de productos de degradación del fibrinógeno predecían un pobre desenlace, independientemente de otras variables, y que el pronóstico empeoraba cuando los niveles de productos de degradación del fibrinógeno aumentaban. En pacientes con trauma craneoencefálico moderado, el tiempo parcial activado de tromboplastina parece tener una mayor relación con el pronóstico del paciente. Los resultados del estudio IMPACT48 muestran que el tiempo de protrombina es un fuerte factor de pronóstico independiente, después del trauma craneoencefálico.
TRATAMIENTO
No existen guías para el tratamiento de la coagulopatía posterior al trauma craneoencefálico.
En general, la terapia de los trastornos de la coagulación debe ser dirigida a tratar la causa que los desencadenó.
Se ha propuesto que los objetivos del tratamiento se deben orientar a combatir la coagulopatía y la lisis de coágulos existentes, a reponer los factores de coagulación y a revertir la hiperfibrinólisis49. El uso de plasma fresco congelado y aféresis de plaquetas, se recomienda en pacientes con sangrado activo. El plasma fresco congelado, como líquido de reanimación en pacientes con puntaje Glasgow de 7o menor50 y en niños con puntajes de Glasgow de 8o menor43, se ha usado para combatir las alteraciones de la coagulación y prevenir la progresión de las lesiones.
El uso de proteína C activada no se ha estudiado en casos de trauma craneoencefálico. En casos de sepsis, se han reportado eventos de sangrado grave posiblemente por una mala selección de los pacientes51.
La administración temprana podría inhibir o acortar el tiempo de la coagulopatía.
El uso de agentes antifbrinolíticos, como la aprotinina (retirada del mercado), el ácido tranexámico y el ácido E-amino caproico, es sujeto de estudio actual en casos de trauma (CRASH-2)52.
Los datos preliminares indican que el factor VII a recombinante (rFVIIa) provee una rápida y exitosa corrección de la coagulopatía en pacientes con trauma craneoencefálico, al limitar el crecimiento del hematoma, reducir la mortalidad y mejorar el desenlace funcional a los 90 días53.
En conclusión, el avance en el conocimiento del proceso de coagulación y su comportamiento en el paciente con trauma craneoencefálico, aumenta las estrategias de tratamiento haciendo énfasis en el manejo temprano de la coagulopatía esperada lo que se vera reflejado en un mejor desenlace neurológico y una disminución en la mortalidad.
REFERENCIAS
1. Hees JR, Laeson JH. The coagulopathy of trauma versus disseminated intravascular coagulation. J Trauma. 2006;60:S12-9. [ Links ]
2. Davie, Ratnoff.Coagulopatía del paciente quirúrgico. Revista Mexicana de Anestesiología. 2004;27:219-30. [ Links ]
3. Rott H, Trobisch H, Kretzschmar E. Use of recombinant factor VIIa in the manegement of acute haemorrhage. Cur Opin Anaesthesiol. 2004;17:159-63. [ Links ]
4. Cosgriff N, Monroe EE, Sauaia A, Kenny-Moynihan M, Burch JM, Galloway B. predicting life-threatening coagulophaty in the massively transfused trauma patient: hypothermia and acidosis revisited. J Trauma. 1997;42:857-62. [ Links ]
5. Meng ZH, Wolberg AS, Monroe DO III, Hoffman M. The effect of temperature and ph on the activity of factor VIIa: implications for trhe effcacy of high-dose factor VIIa in hypothermic and acidotic patients. J Trauma. 2003;55:886-91. [ Links ]
6. Kermode JC, Zheng Q, Milner EP. Marked temperature dependence of the platelet calcium signal induced by human von Willebrand factor. Blood. 1999;94:199-207. [ Links ]
7. Baker SP, ONeill B, Haddon W, Long WB. The injury severity score: A method for describing patients with multiple injuries and evaluating emergency care. J Trauma. 1974;14:187-96. [ Links ]
8. Harhangi BS, Kompanje EJO, Leebeek FWG, Maas AIR. Coagulation disorders after traumatic brain injury. Acta Neurochir (Wien). 2008;150:165-75. [ Links ]
9. Hulka F, Mullins RJ, Frank EH. Blunt brain injury activates the coagulation process. Arch Surg. 1996;131:923-7. [ Links ]
10. Piek J, Chesnut RM, Marshall LF, van Berkum-Clark M, Klauber MR, Blunt BA, et al. Extracranial complications of severe head injury. J Neurosurg. 1992;77:901-7. [ Links ]
11. Giesen PL, Nemerson Y. Tissue factor on the loose. Semen Thromb Hemost. 2000;26:379-84. [ Links ]
12. Scherer RU, Spangenberg P. Procoagulant activity in patients with isolated severe head trauma. Crit Care Med. 1998;26:149-56. [ Links ]
13. Lawson JH, Kalafatis M, Stram S, Mann KG. A model for the tissue factor pathway to thrombin. I. An empirical study. J Biol Chem. 1994;269:357-66. [ Links ]
14. Keimowitz RM, Annis BL. Disseminated intravascular coagulation associated with massive brain injury. J Neurosurg. 1973;39:178-80. [ Links ]
15. Astrup T. Assay and content of tissue thromboplastin in different organs. Thromb Diath Haemorrh. 1965;14:401-6. [ Links ]
16. Goodnight SH, Kenoyer G, Rapaport SI, Patch MJ, Lee JA, Kurze T. Defibrination after brain-tissue destruction: a serious complication of head injury. N Engl J Med. 1974;290:1043-7. [ Links ]
17. Gando S. Disseminated intravascular coagulation in trauma patients. Semin Thromb Hemost. 2001;27:585-92. [ Links ]
18. Pathak A, Dutta S, Marwaha N, Singh D, Varma N, Mathuriya SN. Change in tissue thromboplastin content of brain following trauma. Neurol India. 2005;53:178-82. [ Links ]
19. Mann KG, Butenas S, Brummel K. The dynamics of thrombin formation. Arterioscler Thromb Vasc Biol. 2003;23:17-25. [ Links ]
20. Kaufman HH, Hui KS, Mattson JC, Borit A, Childs TL, HootsWK, et al. Clinicopathological correlations of disseminated intravascular coagulation in patients with head injury. Neurosurgery. 1984;15:34-42. [ Links ]
21. Levi M. Disseminated intravascular coagulation: whats new? Crit Care Clin. 2005;21:449-67. [ Links ]
22. Levi M, Ten Cate H. Disseminated intravascular coagulation. N Engl J Med. 1999;341:586-92. [ Links ]
23. Miner ME, Kaufman HH, Graham SH, Haar FH, Gildenberg PL. Disseminated intravascular coagulation fibrinolytic syndrome following head injury in children: frequency and prognostic implications. J Pediatr. 1982;100:687-91. [ Links ]
24. Nekludov M, Bellander BM, Blombäck M, Wallen HN. Platelet dysfunction in patients with severe traumatic brain injury. J. Neurotrauma. 2008;24:1699-706. [ Links ]
25. Stein SC, Smith DH. Coagulopathy in traumatic brain injury. Neurocrit Care. 2004;1:479-88. [ Links ]
26. Gando S, Nanzaki S, Kemmotsu O. Coagulo fibrinolytic changes after isolated head injury are not different from those in trauma patients without head injury. J Trauma. 1999;46:1070-7. [ Links ]
27. Hess JR, Lawson JH. The coagulopathy of trauma versus disseminated intravascular coagulation. J Trauma 2006;60:S12-9. [ Links ]
28. Jacoby RC, Owings JT, Holmes J, Battistella FD, Gosselin RC, Paglieroni TG. Platelet activation and function after trauma. J Trauma. 2001;51:639-47. [ Links ]
29. Carrick MM, Tyroch AH, Youens CA, Handley T. Subsequent development of thrombocytopenia and coagulopathy in moderate and severe head injury: support for serial laboratory examination. J Trauma. 2005;58:725-30. [ Links ]
30. Engstrom M, Romner B, Schalen W, Reinstrup P. Thrombocytopenia predicts progressive hemorrhage after head trauma. J Neurotrauma. 2005;22:291-6. [ Links ]
31. Parry-Jones BL, Vaughan FL, Miles Cox W. Traumatic brain injury and substance misuse: a systematic review of prevalence and outcomes research (1994-2004). Neuropsychol Rehabil. 2006;16:537-60. [ Links ]
32. Bell A. The Ivy bleeding time. Am J Med Technol. 1958;24: 264-70. [ Links ]
33. Singer AJ, Mynster CJ, Mcmahon BJ. The effect of IM ketorolac tromethamine on bleeding time: a prospective, interventional, controlled study. Am J Emerg Med. 2003;21:441-3. [ Links ]
34. Raffán F, Ramírez FJ, Cuervo JA, Sánchez LF. Tromboelastografía. Rev Col Anes. 2005;33:181-6. [ Links ]
35. Maeda T, Katayama Y, Kawamata T, Aoyama N, Mori T. Hemodynamic depression and microthrombosis in the peripheral areas of cortical contusion in the rat: role of platelet activating factor. Acta Neurochir. 1997;70(Suppl.):102-5. [ Links ]
36. Schwarzmaier S, Kim SW, Trabold R, Plesnila N. Intravascular leukocytes and secondary brain damage after experimental TBI. Presented at the 8th International NeuroTrauma Society (INTS) Meeting. 2006. [ Links ]
37. Auer L. Disturbances of the coagulatory system in patients with severe cerebral trauma. I. Acta Neurochir (Wien). 1978;43:51-9. [ Links ]
38. Cortiana M, Zagara G, Fava S, Seveso M. Coagulation abnormalities in patients with head injury. J Neurosurg Sci. 1986;30:133-8. [ Links ]
39. Hoyt DB. A clinical review of bleeding dilemmas in trauma. Semin Hematol. 2004;41(Suppl.1):40-3. [ Links ]
40. Gómez PA, Lobato RD, Ortega JM, De La Cruz J. Mild head injury: differences in prognosis among patients with a Glasgow Coma Scale score of 13 to 15 and analysis of factors associated with abnormal CT findings. Br J Neurosurg. 1996;10:453-60. [ Links ]
41. Kumura E, Sato M, Fukuda A, Takemoto Y, Tanaka S, Kohama A. Coagulation disorders following acute head injury. Acta Neurochir (Wien). 1987;85:23-8. [ Links ]
42. Schreiber MA, Differding J, Thorborg P, Mayberry JC, Mullins RJ. Hypercoagulability is most prevalent early after injury and in female patients. J Trauma. 2005;58:475-80; discussion 480-71. [ Links ]
43. Keller MS, Fendya DG, Weber TR. Glasgow Coma Scale predicts coagulopathy in pediatric trauma patients. Semin Pediatr Surg. 2001;10:12-6. [ Links ]
44. Stein SC, Spettell C, Young G, Ross SE. Delayed and progressive brain injury in closed-head trauma: radiological demonstration. Neurosurgery. 1993;32:25-30; discussion 30-21. [ Links ]
45. Olson JD, Kaufman HH, Moake J, OGorman TW, Hoots K, Wagner K, Brown CK, Gildenberg PL. The incidence and significance of haemostatic abnormalities in patients with head injuries. Neurosurgery. 1989;24:825-32. [ Links ]
46. Selladurai BM, Vickneswaran M, Duraisamy S, Atan M. Coagulopathy in acute head injury – a study of its role as a prognostic indicator. Br J Neurosurg. 1997;11:398-404. [ Links ]
47. Kushimoto S, Shibata Y, Yamamoto Y. Implications of fibrinogenolysis in patients with closed head injury. J Neurotraum. 2003;20:357-63. [ Links ]
48. Murray GD, Butcher I, McHugh GS, Lu J, Mushkudiani NA, Maas I, Marmarou A, Steyerberg EW. Multivariable prognostic analysis in traumatic brain injury: results from the IMPACT study. J Neurotraum. 2007;24:329-37. [ Links ]
49. Stein SC, Smith DH. Coagulopathy in traumatic brain injury. Neurocrit Care. 2004;1:479-88. [ Links ]
50. May AK, Young JS, Butler K, Bassam D, Brady W. Coagulopathy in severe closed head injury: is empiric therapy warranted? Am Surg. 1997;63:233-6; discussion 236-7. [ Links ]
51. Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, López-Rodríguez A, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699-709. [ Links ]
52. Roberts I. The CRASH-2 trial of an antifibrinolytic agent in traumatic haemorrhage: an international collaboration. Indian J Med Res. 2007;125:5-7. [ Links ]
53. Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, et al.Recombinant activated factor VII for acute intracerebral haemorrhage. N Engl J Med. 2005;352:777-85. [ Links ]
